
Amyotrophic lateral sclerosis (ALS) – Lou Gehrig’disease
DEFINITION
Motor neuron diseases (MND) include a heterogeneous spectrum of inherited and sporadic (no family history) clinical disorders of the upper motor neurons (UMNs), lower motor neurons (LMNs), or a combination of both. Amyotrophic lateral sclerosis (ALS), commonly known as” Lou Gehrig’disease”, is the most common and devastatingly fatal MND among adults. ”ALS is characterized by the degeneration and loss of motor neurons in the spinal cord, brainstem, and brain, resulting in a variety of UMN and LMN clinical signs and symptoms”.
Amyotrophic lateral sclerosis (ALS) is a group of rare neurological diseases that mainly involve the nerve cells (neurons) responsible for controlling voluntary muscle movement. Voluntary muscles produce movements like chewing, walking, and talking. The disease is progressive, meaning the symptoms get worse over time. Currently, there is no cure for ALS and no effective treatment to halt, or reverse, the progression of the disease.
ALS belongs to a wider group of disorders known as motor neuron diseases, which are caused by gradual deterioration (degeneration) and death of motor neurons. Motor neurons are nerve cells that extend from the brain to the spinal cord and to muscles throughout the body. These motor neurons initiate and provide vital communication links between the brain and the voluntary muscles.
Messages from motor neurons in the brain (called upper motor neurons) are transmitted to motor neurons in the spinal cord and to motor nuclei of the brain (called lower motor neurons) and from the spinal cord and motor nuclei of the brain to a particular muscle or muscles.
In ALS, both the upper motor neurons and the lower motor neurons degenerate or die, and stop sending messages to the muscles. Unable to function, the muscles gradually weaken, start to twitch (called fasciculations), and waste away (atrophy). Eventually, the brain loses its ability to initiate and control voluntary movements.
Early symptoms of ALS usually include muscle weakness or stiffness. Gradually all muscles under voluntary control are affected, and individuals lose their strength and the ability to speak, eat, move, and even breathe.
Most people with ALS die from respiratory failure, usually within 3 to 5 years from when the symptoms first appear. However, about 10 percent of people with ALS survive for 10 or more years.
Epidemiology
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disorder of unknown etiology. ALS onset is rare before age 40 and increases with age thereafter. Men are at higher risk than women (ratio 1.3:1). Other than age and gender, the only indisputable risk factor for ALS is genetic susceptibility, with familial cases occurring in about 10% of most case series.
Genetic linkage studies have provided evidence that a mutant form of the gene that codes for Cu/Zn superoxide dismutase1(SOD1), an endogenous free radical scavenger, is important in 15-20% of familial cases. Epidemiologic studies have identified associations of sporadic ALS with work in occupations that involve toxicant exposure.
Environmental toxicants may act against a background of increased genetic susceptibility; however, genetically acquired biochemical defects have not been identified in sporadic ALS patients. Other epidemiologic theories of disease etiology have emphasized the potential role of physical trauma, electrical shock, and vigorous physical exertion, but evidence regarding these factors is inconsistent.
Risk Factor
In 2016 the Centers for Disease Control and Prevention estimated that between 14,000 – 15,000 Americans have ALS. ALS is a common neuromuscular disease worldwide. It affects people of all races and ethnic backgrounds.
There are several potential risk factors for ALS including:
AGE- Although the disease can strike at any age, symptoms most commonly develop between the ages of 55 and 75.
GENDER-. Men are slightly more likely than women to develop ALS. However, as we age the difference between men and women disappears.
RACE-and ethnicity. Most likely to develop the disease are Caucasians and non-Hispanics.
Some studies suggest that military veterans are about 1.5 to 2 times more likely to develop ALS. Although the reason for this is unclear, possible risk factors for veterans include exposure to lead, pesticides, and other environmental toxins. ALS is recognized as a service-connected disease by the U.S. Department of Veterans Affairs.
TYPES OF AMYOTROPHIC LATERAL SCLEROSIS
1-SPORADIC ALS:
The majority of ALS cases (90 percent or more) are considered sporadic. This means the disease seems to occur at random with no clearly associated risk factors and no family history of the disease. Although family members of people with sporadic ALS are at an increased risk for the disease, the overall risk is very low and most will not develop ALS.
2-FAMILIAL (GENETIC) ALS:
About 5 to 10 percent of all ALS cases are familial, which means that an individual inherits the disease from his or her parents. The familial form of ALS usually only requires one parent to carry the gene responsible for the disease. Mutations in more than a dozen genes have been found to cause familial ALS.
About 25 to 40 percent of all familial cases (and a small percentage of sporadic cases) are caused by a defect in a gene known as “chromosome 9 open reading frame 72,” or C9ORF72. Interestingly, the same mutation can be associated with atrophy of the frontal-temporal lobes of the brain causing frontal-temporal lobe dementia. Some individuals carrying this mutation may show signs of both motor neuron and dementia symptoms (ALS-FTD). Another 12 to 20 percent of familial cases result from mutations in the gene that provides instructions for the production of the enzyme copper-zinc superoxide dismutase 1 (SOD1).
Causes of Amyotrophic lateral sclerosis (ALS)
The cause of ALS is not known, and scientists do not yet know why ALS strikes some people and not others. However, evidence from scientific studies suggests that both genetics and environment play a role in the development of ALS.
Genetics
An important step toward determining ALS risk factors was made in 1993 when scientists supported by the National Institute of Neurological Disorders and Stroke (NINDS) discovered that mutations in the SOD1 gene were associated with some cases of familial ALS.
Although it is still not clear how mutations in the SOD1 gene lead to motor neuron degeneration, there is increasing evidence that the gene playing a role in producing mutant SOD1 protein can become toxic.
Since then, more than a dozen additional genetic mutations have been identified, many through NINDS-supported research, and each of these gene discoveries is providing new insights into possible mechanisms of ALS.
The discovery of certain genetic mutations involved in ALS suggests that changes in the processing of RNA molecules may lead to ALS-related motor neuron degeneration. RNA molecules are one of the major macromolecules in the cell involved in directing the synthesis of specific proteins as well as gene regulation and activity.
Other gene mutations indicate defects in the natural process in which malfunctioning proteins are broken down and used to build new ones, known as protein recycling. Still, others point to possible defects in the structure and shape of motor neurons, as well as increased susceptibility to environmental toxins. Overall, it is becoming increasingly clear that a number of cellular defects can lead to motor neuron degeneration in ALS.
In 2011 another important discovery was made when scientists found that a defect in the C9ORF72 gene is not only present in a significant subset of individuals with ALS but also in some people with a type of frontotemporal dementia (FTD). This observation provides evidence for genetic ties between these two neurodegenerative disorders. Most researchers now believe ALS and some forms of FTD are related disorders.
Environmental factors-
In searching for the cause of ALS, researchers are also studying the impact of environmental factors. Researchers are investigating a number of possible causes such as exposure to toxic or infectious agents, viruses, physical trauma, diet, and behavioral and occupational factors.
For example, researchers have suggested that exposure to toxins during warfare, or strenuous physical activity, are possible reasons why some veterans and athletes may be at increased risk of developing ALS.
Although there has been no consistent association between any environmental factor and the risk of developing ALS, future research may show that some factors are involved in the development or progression of the disease.
CLINICAL MANIFESTATION
The onset of ALS can be so subtle that the symptoms are overlooked but gradually these symptoms develop into more obvious weakness or atrophy that may cause a physician to suspect ALS. Some of the early symptoms include:
IMPAIRMENTS RELATED TO THE LMN PATHOLOGY-
- Weakness of the upper extremity (UE), lower extremity (LE), or the weakness of the bulbar muscles. muscle weakness is the cardinal sign of ALS.
- Cervical extensor weakness is typical.
- Sensory pathways are spared for the most part in people with ALS.
- Muscle weakness leads to secondary impairments, including decreased ROM, predisposing the
- patient to joint subluxation, joint contracture(commonly claw hand deformity), tendon
- shortening(Achilles), and adhesive capsulitis.
- FOOT DROP secondary to the distal weakness, and INSTABILITY secondary to the proximal weakness, are common.
- Several factors affect the FATIGUE levels in ALS.
As motor neurons die, the remaining neurons or sprouted neurons are overburdened. weak muscles must work at a higher percentage of their maximal strength to perform the same activity. this hatens the muscle fatigue. - Fasciculations (muscle twitches) in the arm, leg, shoulder, or tongue
- Muscle cramps
- Muscle weakness affecting an arm, a leg, neck or diaphragm.
- Other LMN signs include hyporeflexia,decreased or absent reflex,flaccidity and muscle cramping.
- For many individuals the first sign of ALS may appear in the hand or arm as they experience difficulty with simple tasks such as Buttoning a shirt, writing, or turning a key in a lock. In other cases, symptoms initially affect one of the legs, and people Experience awkwardness when walking or running or they notice that they are tripping or stumbling more often.
IMAPIRMENTS RELATED TO UMN PATHOLOGY-
- UMN loss characterised by spasticity,hyperflexia,clonus,and pathological reflexes, such as a babinski or hoffmann sign, and may also cause muscle weakness.
IMPAIRMENTS RELATED TO BULBAR PATHOLOGY-
- When symptoms begin in the arms or legs, it is referred to as “limb onset” ALS”. Other individuals first notice speech or Swallowing problems, termed “bulbar onset” ALS.
- Regardless of where the symptoms first appear, muscle weakness and atrophy spread to other parts of the body as the disease progresses. Individuals may develop problems with moving, swallowing (dysphagia), speaking or forming words (dysarthria), and breathing (dyspnea).
- Although the sequence of emerging symptoms and the rate of disease progression vary from person to person, eventually individuals will not be able to stand or walk, get in or out of bed on their own, or use their hands and arms.
- Individuals with ALS usually have difficulty swallowing and chewing food, which makes it hard to eat normally and increases the risk of choking. They also burn calories at a faster rate than most people without ALS. Due to these factors, people with ALS tend to lose weight rapidly and can become malnourished.
- Because people with ALS usually retain their ability to perform higher mental processes such as reasoning, remembering, understanding, and problem solving, they are aware of their progressive loss of function and may become anxious and depressed.
- A small percentage of individuals may experience problems with language or decision-making, and there is growing evidence that some may even develop a form of dementia over time.
- Individuals with ALS will have difficulty breathing as the muscles of the respiratory system weaken. They eventually lose the ability to breathe on their own and must depend on a ventilator. Affected individuals also face an increased risk of pneumonia during later stages of the disease. Besides muscle cramps that may cause discomfort, some individuals with ALS may develop painful neuropathy (nerve disease or damage).
SYMPTOMS-
1-ADVANCE SYMPTOMS-
- Muscle weakness
- Muscle Atrophy
- Movement difficulty
- Spasticity
- Excess saliva from reduced swallowing
- Thicker saliva may sometimes be difficult to clear from the chest or throat due to weakening of muscles
- Excess yawning
- Emotionality
- Difficulties with concentration, planning and use of language
- Fronto-temporal dementia – this affects their ability to problem solve and respond to new situations[
- Breathing difficulties, such as shortness of breath
END STAGE SYMPTOMS-
- Increasing body paralysis
- Significant shortness of breath
- 2-SECONDERY SYMPTOMS-
These are not directly caused by the disease but are related to the stress of living with it. - Depression
- Insomnia
- Anxiety
DIAGNOSIS:
Amyotrophic lateral sclerosis (ALS) is difficult to diagnose early because it may mimic several other neurological diseases. Tests to rule out other conditions may include:
ELECTOMYOGRAM (EMG)- During an EMG, your doctor inserts a needle electrode through your skin into various muscles. The test evaluates the electrical activity of your muscles when they contract and when they’re at rest.
Abnormalities in muscles seen in an electromyogram can help doctors diagnose ALS, or determine if you have a different muscle or nerve condition that may be causing your symptoms. It can also help guide your exercise therapy.
NERVE CONDUCTION STUDY(NCS)-This study measures your nerves’ ability to send impulses to muscles in different areas of your body. This test can determine if you have nerve damage or certain muscle diseases.
MAGNETIC RESONANCE IMAGINE (MRI)- Using radio waves and a powerful magnetic field, an MRI produces detailed images of your brain and spinal cord. An MRI can spot spinal cord tumors, herniated disks in your neck or other conditions that may be causing your symptoms.
BLOOD AND URINE TEST- Analyzing samples of your blood and urine in the laboratory may help your doctor eliminate other possible causes of your signs and symptoms.
SPINAL TAP (lumbar puncture)- Sometimes a specialist may remove a sample of your spinal fluid for analysis. A specialist inserts a small needle between two vertebrae in your lower back and removes a small amount of cerebrospinal fluid for testing in the laboratory.
MUSCLE BIOPSY- If your doctor believes you may have a muscle disease rather than ALS, you may undergo a muscle biopsy. While you’re under local anesthesia, a small portion of your muscle is removed and sent to a lab for analysis.
TREATMENT AND PREVENTION
Physical therapy can help people with ALS manage pain and address mobility issues.
A physical therapist can provide help and information with:
low-impact exercises to enhance cardiovascular fitness and overall well-being
mobility aids, such as walkers and wheelchairs
devices to make life easier, such as ramps
Occupational therapy can help a patient maintain their independence for longer by:
helping patients choose adaptive equipment and assistive technologies to help them keep up their daily routines
train them in ways to compensate for hand and arm weaknesses
Breathing therapy may be needed in time, as the respiratory muscles get weaker.
- Breathing devices- can help the patient breathe better at night. Some patients may need mechanical ventilation. One end of a tube is connected to a respirator, while the other end is inserted into the windpipe through a surgically-created hole in the neck, or tracheostomy.
- Speech therapy- is useful when ALS begins to make it harder to talk. Speech therapists can help by teaching adaptive techniques. Other methods of communication include writing and computer-based communications equipment.
- Nutritional support is important, as difficulty with swallowing can make it hard to get enough nutrients. Nutritionists can advise on preparing nutritious meals that are easier to swallow. Suction devices and feeding tubes may help.
There is no cure for ALS, so treatment aims to alleviate symptoms, prevent unnecessary complications, and slow the rate of disease progression. - ALS can cause a range of physical, mental, and social changes, so a team of specialists will often help patients manage their symptoms and care, improve their qualify of life, and prolong survival.
- Riluzole (Rilutek) was approved for ALS treatment by the Food and Drug Administration (FDA) in 1995, and it appears to slow the progression of the disease. It may work by reducing the body’s levels of glutamate, an excitotoxin that has been linked to neuronal damage.
- In May 2017, Radicava (Edaravone) was approved to treat ALS. It may slow the decline in physical function by one third.
A number of research projects are looking at ways to use new and existing drugs to treat different aspects of ALS. Doctors can also prescribe medications to treat the different symptoms.
PHYSICAL THERAPY INTERVENTION
Physiotherapy helps maintain movement and function when someone is affected by injury, illness or disability. This is achieved through movement and exercise, manual therapy, education and advice. Although physiotherapy can’t reverse the effects of
MND, it can help you to stay mobile and comfortable for as long as possible.
Exercise can also help to:
• keep you mobile for as long as possible by preventing muscles and joints from
becoming stiff
• maintain maximum range of movement (ROM) of joints
• maintain comfort and reduce problems associated with muscle weakness and
stiff joints
• maintain circulation through active muscle movement.
Breathing exercises can help with chest clearance and reducing the chance of chest infections
1. Carry out a full assessment to check your mobility, range of movement of joints and muscles, strength of muscles and discuss / see any particular difficulties you’re experiencing.
2. Advise on and provide walking aids that will help to keep your mobility as long and as safely as possible.
3. Relieve pain and tension from muscle cramps with gentle passive stretches and massage.
4. Devised a tailored exercise programme to strenghten the muscles that have not been affected by MND – some exercises may be active and some active-assistsed meaning the physio helps you to carry out the exercises.
5. Passive exercises to help move the joints you find difficult, reduce tension and aid circulation.
6.Teach stretches that can be carried out on your own to help keep muscles from becoming tight.
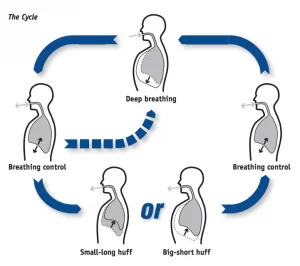
7. Help posture and positioning in the later stages and show carers how to use pillows to keep individuals as comfortable as possible and ensure breathing is maximised.8. Teach breathing exercises and check chest clearing techniques. The Active Cycle of Breathing Techniques is shown in the picture.
POSITIONING
ACTIVE CYCLE OF BREATHING TECHNIQUE (ACBT)
9. Teach how to pace your exercises and stop you overdoing it and being in pain.
10. Advise on pain relief and using a TENS machine, heat or ice.
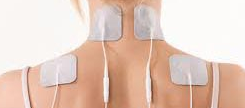
Author: Nitesh Patel - Physiotherapist
Physiotherapist in Samarpan Physiotherapy Clinic Ahmedabad Bapunagar Amaraiwadi Vastral Mobile Physiotherapy Clinic Dr. Nitesh Patel ( Physiotherapist ) : Mo No : 09898607803


2 Comments