
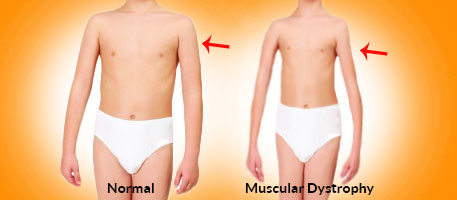
Muscular Dystrophy
What is a Muscular Dystrophy?
Muscular dystrophy disease is genetic, and consequently, a history in the family increases the chance of an individual developing the disease.
Symptoms of Muscular Dystrophy
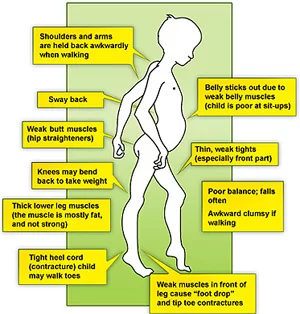
→ Initial symptoms
A waddling gait
Pain and stiffness in the muscles
Difficulty with running and jumping
Walking on toes
Difficulty sitting up or standing
Learning disabilities, such as developing speech later than usual
Frequent falls
Gower’s sign positive
→ Later symptoms
Inability to walk
A shortening of muscles and tendons, further limits movement
Breathing problems can become so severe that assisted breathing is necessary
The curvature of the spine can be caused if muscles are not strong enough to support its structure
The muscles of the heart can be weakened, leading to cardiac problems
Difficulty swallowing – this can cause aspiration pneumonia, and a feeding tube is sometimes necessary
Causes
→ Muscular dystrophy is caused by mutations on the X chromosome. Each version of muscular dystrophy is due to a different set of mutations, but all prevent the body from producing dystrophin. Dystrophin is a protein essential for building and repairing muscles.
→ Dystrophin is part of an incredibly complex group of proteins that allow muscles to work correctly. The protein helps anchor various components within muscle cells together and links them all to the sarcolemma – the outer membrane.
→ If dystrophin is absent or deformed, this process does not work correctly, and disruptions occur in the outer membrane. This weakens the muscles and can also actively damage the muscle cells themselves.
Types of Muscular Dystrophy
(1) Becker muscular dystrophy:
→ Becker muscular dystrophy (BMD) is a less severe variant of Duchenne muscular dystrophy and is caused by the production of a truncated, but partially functional form of dystrophin. Survival is usually into old age and affects only boys (with extremely rare exceptions
(2) Congenital muscular dystrophy:
→ Age at onset is birth, the symptoms include general muscle weakness and possible joint deformities, the disease progresses slowly, and the lifespan is shortened. Congenital muscular dystrophy includes several disorders with a range of symptoms. Muscle degeneration may be mild or severe. Problems may be restricted to skeletal muscle, or muscle degeneration may be paired with effects on the brain and other organ systems.
→ Several forms of congenital muscular dystrophies are caused by defects in proteins thought to have some relationship to the dystrophin-glycoprotein complex and to the connections between muscle cells and their surrounding cellular structure. Some forms of congenital muscular dystrophy show severe brain malformations, such as lissencephaly and hydrocephalus
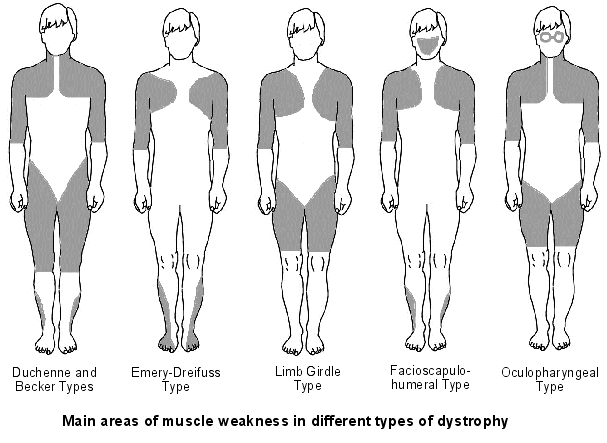
(3) Duchenne muscular dystrophy(DMD):
→ Duchenne muscular dystrophy (DMD) is the most common childhood form of muscular dystrophy; it generally affects only boys (with extremely rare exceptions), becoming clinically evident when a child begins walking. By age 10, the child may need braces for walking, and by age 12, most patients are unable to walk. Lifespans range from 15 to 45, though a few exceptions occur. Researchers have identified the gene for the protein dystrophin, which when absent, causes DMD. Since the gene is on the X chromosome, this disorder affects primarily males, and females who are carriers have milder symptoms. Sporadic mutations in this gene occur frequently.
→ Dystrophin is part of a complex structure involving several other protein components. The “dystrophin-glycoprotein complex” helps anchor the structural skeleton (cytoskeleton) within the muscle cells, through the outer membrane (sarcolemma) of each cell, to the tissue framework (extracellular matrix) that surrounds each cell. Due to defects in this assembly, contraction of the muscle leads to disruption of the outer membrane of the muscle cells and eventual weakening and wasting of the muscle.
(4) Distal muscular dystrophy:
→ Distal muscular dystrophies age at onset is about 20 to 60 years; symptoms include weakness and wasting of muscles of the hands, forearms, and lower legs; progress is slow and not life-threatening.
→ Miyoshi myopathy, one of the distal muscular dystrophies, causes initial weakness in the calf muscles and is caused by defects in the same gene responsible for one form of limb-girdle muscular dystrophy
(5) Emery–Dreifuss muscular dystrophy:
→ Emery–Dreifuss muscular dystrophy patients normally present in childhood and the early teenage years with contractures. Clinical signs include muscle weakness and wasting, starting in the distal limb muscles and progressing to involve the limb-girdle muscles. Most patients also suffer from cardiac conduction defects and arrhythmia.
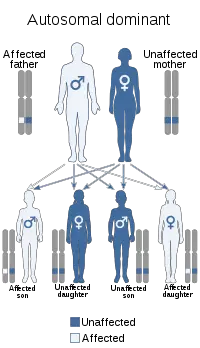
→ The three subtypes of Emery–Dreifuss MD are distinguishable by their pattern of inheritance: X-linked, autosomal dominant, and autosomal recessive. The X-linked form is the most common. Each type varies in prevalence and symptoms. The disease is caused by mutations in the LMNA gene, or more commonly, the EMD gene. Both genes encode protein components of the nuclear envelope. However, how these mutations cause pathogenesis is not well understood.
(6) Facioscapulohumeral muscular dystrophy:
→ Facioscapulohumeral muscular dystrophy (FSHD) initially affects the muscles of the face, shoulders, and upper arms with progressive weakness.
→ Symptoms usually develop in early adulthood (late teens); affected individuals become severely disabled. The pattern of inheritance is autosomal dominant, though a number of spontaneous mutations occur. Two defects are needed for FSHD, which for the first time provides a unifying theory for the underlying genetics of FSHD.
FSHD occurs both in males and females.
(7) Limb-girdle muscular dystrophy(LGMD):
→ Limb-girdle muscular dystrophy (LGMD) affects both boys and girls. LGMDs all show a similar distribution of muscle weakness, affecting both upper arms and legs. Many forms of LGMD have been identified, showing different patterns of inheritance (autosomal recessive vs. autosomal dominant). In an autosomal recessive pattern of inheritance, an individual receives two copies of the defective gene, one from each parent. The recessive LGMDs are more frequent than the dominant forms and usually have childhood or teenaged onset. The dominant LGMDs usually show adult onset. Some of the recessive forms have been associated with defects in proteins that make up the dystrophin-glycoprotein complex.[9] Though a person normally leads a normal life with some assistance, in some extreme cases, death from LGMD occurs due to cardiopulmonary complications
(8) Myotonic muscular dystrophy:
→ Myotonic muscular dystrophy is an autosomal dominant condition that presents with myotonia (delayed relaxation of muscles), as well as muscle wasting and weakness. Myotonic MD varies in severity and manifestations and affects many body systems in addition to skeletal muscles, including the heart, endocrine organs, and eyes.
Myotonic MD type 1 (DM1) is the most common adult form of muscular dystrophy. It results from the expansion of a short (CTG) repeat in the DNA sequence of the myotonic dystrophy protein kinase gene. Myotonic muscular dystrophy type 2 (DM2) is rarer and is a result of the expansion of the CCTG repeat in the zinc finger protein 9 gene.
(9) Oculopharyngeal muscular dystrophy:
→ Oculopharyngeal MD’s age at onset is 40 to 70 years; symptoms affect muscles of eyelids, face, and throat followed by pelvic and shoulder muscle weakness; it has been attributed to a short repeat expansion in the genome which regulates the translation of some genes into functional proteins.
Diagnosis
→ Enzyme assay – damaged muscles produce creatine kinase (CK). Elevated levels of CK in the absence of other types of muscle damage could suggest muscular dystrophy.
→ Genetic testing – as genetic mutations are known to occur in muscular dystrophy, these changes can be screened for.
→ Heart monitoring – electrocardiography and echocardiograms can detect changes in the musculature of the heart. This is especially useful for the diagnosis of myotonic muscular dystrophy.
→ Lung monitoring – checking lung function can give additional evidence.
→ Electromyography – a needle is placed into the muscle to measure the electrical activity. The results can show signs of muscle disease.
→ Biopsy- removing a portion of the muscle and examining it under a microscope can show the tell-tale signs of muscular dystrophy.
Treatment of Muscular Dystrophy
→ Drugs – Corticosteroids – although this type of medication can help increase muscle strength and slow progression, their long-term use can weaken the bone and increase weight gain
Heart medications – if muscular dystrophy impacts the heart, beta blockers and angiotensin-converting enzyme (ACE) inhibitors may be useful.
Physiotherapy treatment of Muscular Dystrophy
→ General exercises – a range of motion and stretching exercises can help the movement of the limbs as muscles and tendons shorten.

→ low-impact aerobic exercises such as walking and swimming can also help slow the disease’s progression

→ Breathing assistance – as the muscles used for breathing become weaker, it may be necessary to use devices to help improve oxygen delivery through the night.
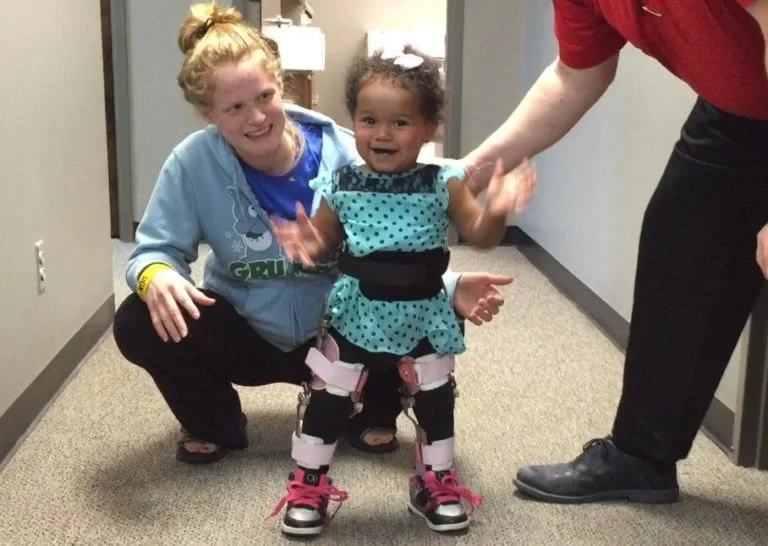
→ Mobility aids – canes, wheelchairs, and walkers.
→ Braces – these keep muscles and tendons stretched and help slow their shortening. They also give added support to the user when moving.
Author: Nitesh Patel - Physiotherapist
Physiotherapist in Samarpan Physiotherapy Clinic Ahmedabad Bapunagar Amaraiwadi Vastral Mobile Physiotherapy Clinic Dr. Nitesh Patel ( Physiotherapist ) : Mo No : 09898607803



2 Comments