Neuromuscular Junction
Definition
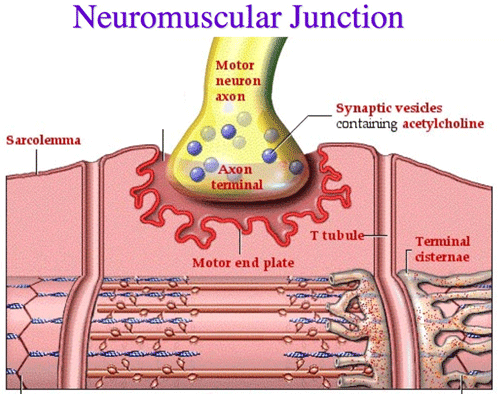
A neuromuscular junction, sometimes referred to as a myoneural junction, is the chemical synapse formed between a motor neuron and a muscle fiber.
Through communication with the muscle fiber, the motor neuron is able to deliver a signal that causes the muscle to contract.
The terminal end of a motor neuron and a muscle (skeletal, smooth, or cardiac) form the neuromuscular junction (NMJ), which is a synaptic connection. It is the location where the action potential is transferred from the nerve to the muscle. It is also a site of action for a range of drugs as well as a place for a number of ailments.
Introduction
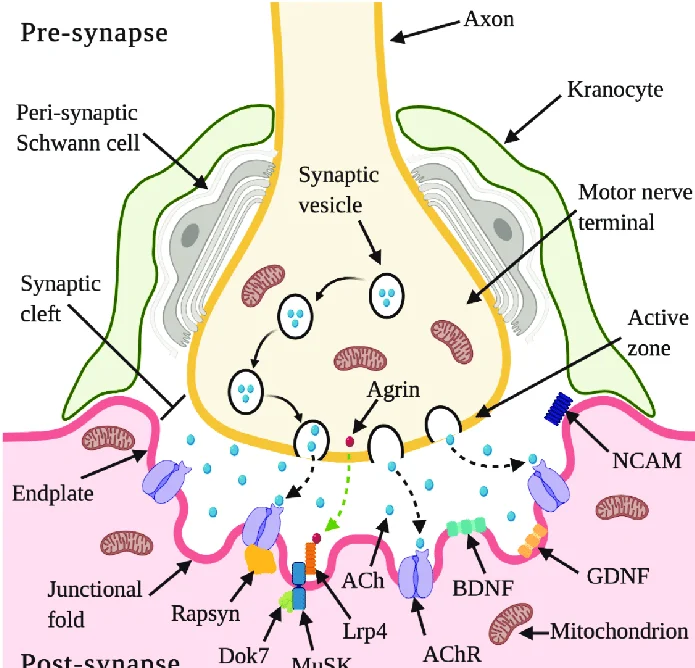
Muscles require innervation to be able to operate, or even just to maintain their tone and avoid atrophy. The neuromuscular system, where they collaborate with muscles, connects the nerves of the peripheral and central nervous systems. The presynaptic terminal of a motor neuron activates voltage-gated calcium channels, allowing calcium ions to enter the neuron and starting synaptic transmission at the neuromuscular junction. Synaptic vesicle fusion with the cell membrane and subsequent neurotransmitter release from the motor neuron into the synaptic cleft is triggered by calcium ions binding to sensor proteins (synaptotagmins) on synaptic vesicles.
Acetylcholine is a small-molecule neurotransmitter that is released by motor neurons in vertebrates. Acetylcholine binds to nicotinic acetylcholine receptors (nAChRs) on the cell membrane of the muscle fiber, commonly known as the sarcolemma. Ionotropic receptors, or ligand-gated ion channels, include nAChRs. The depolarization of the muscle fiber that might occur as a result of ACh binding to the receptor can eventually lead to muscular contraction.
Neuromuscular junction diseases can be influenced by both hereditary and autoimmune causes. Mutated structural proteins that make up the neuromuscular junction can result in genetic illnesses like Congenital Myasthenic Syndrome, whereas autoimmune diseases like Myasthenia Gravis happen when antibodies are generated against nicotinic acetylcholine receptors on the sarcolemma.
Issues of Concern
Through a variety of methods that may affect the presynaptic, synaptic, or postsynaptic regions of the NMJ, diseases of the NMJ cause muscular weakness. Lambert-Eaton syndrome (LES), myasthenia gravis (MG), and botulism are the three primary illnesses that affect the NMJ.
Structure and function
Motor Endplate
- Quantal transmission
Presynaptic motor axons end at the neuromuscular junction 30 nanometers from the cell membrane or sarcolemma of a muscle fiber. The postjunctional folds on the sarcolemma at the junction enhance the surface area of the structure confronting the synaptic cleft. The motor endplate is made up of these postjunctional folds and is densely packed with 10,000 nicotinic acetylcholine receptors (nAChRs) per micrometer2. The terminal boutons (also known as presynaptic terminals) that the presynaptic axons produce project toward the postjunctional folds of the sarcolemma.
There are around 300,000 vesicles with an average dimension of 0.05 micrometers inside each motor nerve terminal in the frog. Acetylcholine can be found in the vesicles. Some of these vesicles are positioned in active zones near to the neuron membrane in clusters of fifty. A micrometre separates active zones on average. A meshwork of acetylcholinesterase (AChE) with a density of 2,600 enzyme molecules/micrometer2 is present in the 30-nanometer gap between the nerve terminal and endplate, and it is maintained in place by the structural proteins dystrophin and rapsyn. The receptor tyrosine kinase protein MuSK, a signalling protein involved in the formation of the neuromuscular junction, is also present. Rapsyn additionally keeps this junction in place.
In a resting junction, one of the synaptic vesicles randomly fuses with the cell membrane of the presynaptic neuron around once every second. SNARE proteins mediate this process. Exocytosis, which occurs as a result of fusion, causes the vesicle’s 7,000–10,000 cholinergic molecules to be released into the synaptic cleft. As a result, quantal packets of acetylcholine are released during exocytosis. The acetylcholine quantum diffuses across the acetylcholinesterase meshwork, occupying every binding site on the enzyme along its route due to the high local transmitter concentration.
A depolarization of about 0.5 mV known as a miniature endplate potential (MEPP) results from sodium ions moving into the endplate after the acetylcholine that reaches the endplate activates around 2,000 acetylcholine receptors and opens their ion channels. The acetylcholinesterase is ready to disintegrate the acetylcholine that has been released from the receptors by the time it is released from the receptors, which takes around 0.16 ms.
The time between the nerve impulse entering the motor nerve terminals and the endplate’s initial response, when the motor nerve is activated, is just 0.5–0.8 msec. When the motor nerve action potential reaches the presynaptic neuron terminal, voltage-dependent calcium channels open, allowing Ca2+ ions to enter the cytosol of the presynaptic neuron.
Numerous neurotransmitter-containing vesicles fuse with the presynaptic neuron’s cell membrane as a result of this Ca2+ influx, releasing their cholinergic quanta by exocytosis. Endplate potential (EPP) refers to the depolarization of the endplate caused by the released acetylcholine. When ACh binds to nicotinic acetylcholine receptors (nAChR) at the motor end plate, an influx of sodium ions results, which is how the EPP is carried out. The voltage-gated sodium channels in the T-tubules (transverse tubules) allow the action potential to move down the sarcolemma and into the muscle fibre as a result of the influx of sodium ions, which causes the EPP (depolarization).
Action potentials are conducted down the T-tubules, stimulating the opening of voltage-gated Ca2+ channels in the sarcoplasmic reticulum that are mechanically connected to Ca2+ release channels. In order to promote contraction, the Ca2+ then diffuses from the sarcoplasmic reticulum to the myofibrils. Therefore, the endplate potential is in charge of establishing an action potential in the muscle fibre that causes muscular contraction. Each quantum of acetylcholine reaches the endplate at millimolar concentrations, which is high enough to bind with a receptor with a low affinity, which then quickly releases the bound transmitter. This is the reason why the transmission from neuron to muscle is so quick.
Acetylcholine receptors
- Ion channel-linked receptor
- Ions
- Ligand (such as acetylcholine)
- The receptor’s ion channel opens when ligands bind to it, allowing ions to pass through the cell membrane.
- In vertebrates and certain invertebrate creatures, acetylcholine, a neurotransmitter, is involved in the activation of muscle tissue. It is produced from dietary choline and acetyl-CoA (ACoA). The nicotinic acetylcholine receptor (nAChR), a ligand-gated ion channel, is the acetylcholine receptor subtype that is present in the neuromuscular junction of skeletal muscles in vertebrates.
- This receptor’s subunits each have a distinctive “cys-loop” that is made up of a cysteine residue, 13 amino acid residues, and another cysteine residue. The “cys-loop” receptor, which may bind acetylcholine and other ligands, is created when the two cysteine residues form a disulfide connection. Only eukaryotes have these cys-loop receptors, while prokaryotes have ACh receptors that have comparable characteristics. Some animals, such as crayfish and fruit flies, employ a glutamatergic neuromuscular junction rather than a cholinergic one.
AChRs create heteropentamers at the skeletal neuromuscular junction that contain two a, one ß, one? and one d subunits.
A conformational shift at the interface with the second AChR subunit results from the binding of a single ACh ligand to one of the ACh receptor subunits. The second subunit’s affinity for a second ACh ligand is boosted as a result of this conformational shift. Due to this cooperative binding, AChRs have a sigmoidal dissociation curve. ACh that may otherwise be lost by cholinesterase hydrolysis or diffusion is kept in the synapses thanks to the existence of the inactive, intermediate receptor form with a single-bound ligand. A prolonged post-synaptic response may result from the persistence of these ACh ligands in the synaptic cleft.
Development
Both the terminal of the motor neuron and the central portion of the muscle cell must signal for the formation of the neuromuscular junction. Acetylcholine receptors (AChRs) are produced by muscle cells during development and are expressed in the central regions through a process known as repatterning. Once more, it is believed that MuSK kinase and a heparin proteoglycan work together to stabilize the buildup of AChR in the myocyte’s core regions. Because MuSK is a receptor tyrosine kinase, it activates cellular signalling by attaching phosphate molecules to self-regulatory structures like tyrosines and other cytoplasmic targets. When activated by its ligand again, which results in the “clustering” of acetylcholine receptors, MuSK signals via the proteins “Dok-7” and “rapsyn”.
The release of ACh by growing motor neurons results in postsynaptic potentials in the muscle cell that help to stabilize and localize the growing neuromuscular junction.
Mouse “knockout” research helped to partially illustrate these results. The neuromuscular junction does not develop in mice lacking either agrin or MuSK. Furthermore, neither acetylcholine receptor clusters nor neuromuscular connections were formed in mice lacking Dok-7.
Most research on the formation of neuromuscular connections uses model species like rats. In addition, utilizing human embryonic stem cells and somatic muscle stem cells, an all-human neuromuscular junction was developed in vitro in 2015.
In this paradigm, optogenetic activation of presynaptic motor neurons causes synaptically linked muscle fibers to contract in response to light stimulation.
Toxins that affect the neuromuscular junction
Nerve gases
AChE is inactivated as a result of the binding and phosphorylation of AChE by nerve gases. Muscle cells contract continuously as a result of the buildup of ACh in the synaptic cleft, which has severe consequences like paralysis and death within minutes of exposure.
Botulinum toxin injected into a human face
By interacting with SNARE proteins, botulinum toxin (also known as botulinum neurotoxin; commercially available under the trade name Botox) prevents the release of acetylcholine at the neuromuscular junction. Through the process of endocytosis, this toxin enters the nerve terminal and then cleaves the SNARE proteins, preventing the ACh vesicles from joining with the intracellular membrane. This causes a brief flaccid paralysis and localized chemical denervation in the afflicted striated muscle. About two weeks following the injection, the suppression of ACh release begins to take effect. The neuronal activity starts to partially restore function three months after the inhibition takes place, and total neuronal function is resumed six months afterward.
Tetanus toxin
Tetanus is a disease condition brought on by the tetanus toxin, also known as tetanospasmin, which is a powerful neurotoxin produced by Clostridium tetani. This toxin has an estimated LD50 of 1 ng/kg, making it the second-deadliest toxin in the world (after botulinum toxin D). By adhering to and endocytosing into the presynaptic nerve terminal and interfering with SNARE proteins, it works very similarly to botulinum neurotoxin. Tetanospasmin varies from botulinum neurotoxin in a number of ways, most notably in its final form, where it induces spastic paralysis rather than the flaccid paralysis seen with botulinum neurotoxin.
Latrotoxin
The release of acetylcholine from the presynaptic cell is another effect of the latrotoxin (a-Latrotoxin) found in the venom of widow spiders on the neuromuscular junction. The IP3/DAG pathway is activated by attaching to receptors on the presynaptic cell, and calcium is released from intracellular storage, and pores are formed, allowing calcium ions to enter the system directly.
Either process raises calcium levels in the presynaptic cell, which triggers the release of acetylcholine-containing synaptic vesicles. If left untreated, latrotoxin can result in death and paralysis as well as discomfort, muscular tightness, and paralysis.
Snake venom
Snake venom functions as a toxin at the neuromuscular junction that can induce paralysis and weakening. Both presynaptic and postsynaptic neurotoxins can be produced by venoms.
Presynaptic areas of the neuromuscular junction are impacted by presynaptic neurotoxins, also referred to as ß-neurotoxins. The bulk of these neurotoxins work by preventing the entry of neurotransmitters into the synaptic junctions between neurons, including acetylcholine. However, it is also known that certain of these poisons enhance the release of neurotransmitters. The neuromuscular blockade caused by those that restrict neurotransmitter release stops signalling molecules from getting to their postsynaptic target receptors. The result is that the person who was bitten by the snake has severe weakness. These neurotoxins are not adequately countered by anti-venoms. Many of the damaged nerve terminals exhibit evidence of permanent physical damage after being exposed to these toxins for an hour, leaving them devoid of any synaptic vesicles.
A-neurotoxins sometimes referred to as postsynaptic neurotoxins, work in opposition to presynaptic neurotoxins by interacting with the postsynaptic acetylcholine receptors. This stops the receptors on the postsynaptic cell from interacting with the acetylcholine produced by the presynaptic terminal. This effectively prevents the opening of sodium channels connected to these acetylcholine receptors, causing a neuromuscular blockade that is comparable to the effects of presynaptic neurotoxins. As a result, the muscles active in the affected connections become paralysed. Postsynaptic neurotoxins are more susceptible to anti-venoms than presynaptic neurotoxins, which speed up the toxin’s separation from the receptors and eventually reverse paralysis. In research looking at the orientation of antibodies towards the harmed acetylcholine receptors in patients with myasthenia gravis, as well as studies looking at acetylcholine receptor density and turnover, these neurotoxins provide experimental and qualitative support.
Diseases
Neuromuscular junction disease
The phrase “neuromuscular diseases” refers to any condition that affects the synaptic communication between a motor neuron and a muscle cell. These illnesses range in severity and fatality, and they can be inherited or acquired. In general, autoimmune diseases or mutations are the main causes of the majority of these diseases. In the case of neuromuscular illnesses, autoimmune disorders frequently involve humoral and B cell mechanisms and are characterized by the inappropriate production of an antibody against a motor neuron or a muscle fibre protein that influences synaptic signalling or transmission.
Autoimmune
Myasthenia gravis
In 80% of instances of myasthenia gravis, the body produces antibodies against the acetylcholine receptor (AchR), whereas, in 0%–10% of cases, the antibodies are directed towards the postsynaptic muscle-specific kinase (MuSK). Low-density lipoprotein receptor-related protein 4 is the target of IgG1 in seronegative myasthenia gravis, which functions as a competitive inhibitor of its ligand, preventing the ligand from binding to its receptor. If seronegative myasthenia gravis responds to conventional treatments is unknown.
Neonatal MG
One in eight infants born to moms with myasthenia gravis (MG) is affected by the autoimmune condition known as neonatal MG. AChR antibodies can travel across the placenta and pass MG from the mother to the fetus.
Fetal akinesia, or the absence of fetal movement, as well as weakness that reacts to anticholinesterase drugs, are symptoms of this condition at birth. The duration of this sickness in this form is only three months. Neonatal MG, however, can occasionally cause further medical issues, such as arthrogryposis and even fetal demise. These problems can persist until the 33rd week of gestation and are believed to start when maternal AChR antibodies are directed toward the fetal AChR. The e subunit takes the place of the AChR subunit.
Lambert-Eaton myasthenic syndrome
An autoimmune condition known as Lambert-Eaton myasthenic syndrome (LEMS) affects the presynaptic region of the neuromuscular junction. A distinctive trio of symptoms, including proximal muscular weakness, autonomic dysfunction, and areflexia, can identify this uncommon condition. Pathogenic autoantibodies that target P/Q-type voltage-gated calcium channels result in proximal muscle weakness by reducing the release of acetylcholine from motor nerve terminals on the presynaptic cell. Men’s erectile dysfunction, constipation, and, most frequently, dry mouth are a few manifestations of autonomic dysfunction brought on by LEMS. Dry eyes and different sweating patterns are less frequent dysfunctions. Reduced tendon reflexes are a symptom of areflexia, which may temporarily go away after some exercise. A small-cell lung carcinoma (SCLC) is frequently found in 50–60% of individuals with LEMS who also have an associated tumor. Voltage-gated calcium channel expression is also seen in this kind of tumor. LEMS frequently coexists with myasthenia gravis.
The initial step in treating LEMS is to provide 3,4-aminopyridine, which lengthens the duration that voltage-gated calcium channels remain open after blocking voltage-gated potassium channels. This increases the compound muscular action potential and muscle strength. Under an extended access program, 3,4-diamino pyridine therapy is provided without charge to qualified LEMS patients in the US. If 3,4-diaminopyridine is ineffective, other therapies include the use of prednisone and azathioprine.
Neuromyotonia
As with Isaac’s syndrome, neuromyotonia (NMT) differs from many other disorders that affect the neuromuscular junction. NMT results in motor nerve hyperexcitation rather than muscle weakening. The downregulation of voltage-gated potassium channels by NMT results in prolonged depolarizations, which increase neurotransmitter release and repeated firing. This increase in firing rate causes more active transmission, which in turn causes the afflicted person to have higher muscle activity. Due to its connections with autoimmune symptoms in the sick person, NMT is likewise thought to have an autoimmune origin.
Genetic
Congenital myasthenic syndromes
Congenital myasthenic syndromes (CMS) behave very similarly to both MG and LEMS, with the main distinction being that CMS has genetic roots as opposed to both disorders. These syndromes are disorders that affect the presynaptic, synaptic, and postsynaptic proteins at the neuromuscular junction and are specifically brought on by mutations, usually recessive, in 1 of at least 10 genes. These mutations often impact the e-subunit of the AChR, changing the receptor’s kinetics and expression.
A loss of function in the subunit may result from a single nucleotide change or deletion. The expression of CMS can also result from other mutations, including those that alter the enzymes acetylcholinesterase and acetyltransferase, the latter of which is especially linked to episodic apnea. These disorders might manifest at various points during a person’s life. They may develop during the fetal phase, leading to fetal akinesia, or the perinatal stage, when disorders including arthrogryposis, ptosis, hypotonia, ophthalmoplegia, and difficulty breathing or eating may be seen. They could also become active throughout adolescence or adulthood, resulting in slow-channel syndrome in the affected person.
Similar to other neuromuscular illnesses, certain subtypes of CMS (postsynaptic fast-channel CMS) can be treated. The first-line medication for LEMS, 3,4-aminopyridine, is being developed in the US as an orphan medication for CMS and is free of charge to qualified patients through an extended access program.
Summary
To summarize, The neuromuscular junction is a sort of synapse that, in its most basic form, is where neuronal impulses from the brain or spinal cord connect with skeletal muscle fibers to cause their contraction.
Muscle contraction, which can result in movement, is brought about by the simultaneous activation of several muscle fibers. Therefore, a crucial element in the body’s capacity to generate and regulate movement is the neuromuscular junction. Interestingly, the neuromuscular junction’s functions proceed at rates that make motions possible without noticeable lag or delay.
FAQs
What is the neuromuscular junction?
The terminal end of a motor neuron and a muscle (skeletal, smooth, or cardiac) form the neuromuscular junction (NMJ), which is a synaptic connection. It is the place from which the nerve sends the action potential to the muscle.
What is the neuromuscular function?
The capacity for producing regulated movement through synchronized muscle activation is known as neuromuscular control. 27. It is the efferent motor response to sensory information from the vestibular, visual, and proprioceptive systems of the somatosensory (proprioceptive and kinesthetic) system.
What is the first step of the NMJ?
Signaling Processes at Neuromuscular Junctions
The signal first moves down the motor neuron to the presynaptic axon terminal from the axon terminal of the preceding neuron. Calcium channels in the membrane become active and open as a result, enabling calcium ions to enter the cell.
What are the four elements of neuromuscular control?
Joint proprioception and kinesthesia, dynamic stability, preparatory and reactionary muscle characteristics, and conscious and unconscious functional motor patterns are four factors that are essential for regaining neuromuscular control and functional stability.
What is the function of acetylcholine?
Acetylcholine has an impact on a variety of physiological functions, including gut peristalsis, glandular secretion, blood pressure control, heart contractions, and blood flow. Acetylcholine is often an excitatory mediator.
What are neuromuscular skills?
The neuromuscular system may functionally govern and drive movements when sensory feedback, reflex activity, central motor drive, muscle recruitment pattern, muscular excitation-contraction coupling, and energy are well integrated, coordinated, and used.
What are neuromuscular signs?
Neuromuscular diseases symptoms
Muscle weakness
Atrophy of muscle.
Muscle spasms.
Muscle spasticity (stiffness), which leads to skeletal or joint Abnormalities later on.
Muscular ache.
Breathing problems.
Swallowing problems.