
Huntington’s disease (HD)
Overview
Huntington’s disease (HD) is a rare genetic disorder that causes the progressive breakdown of nerve cells in the brain. It is also known as Huntington’s chorea because one of the most characteristic symptoms of the disease is involuntary movements, or chorea.
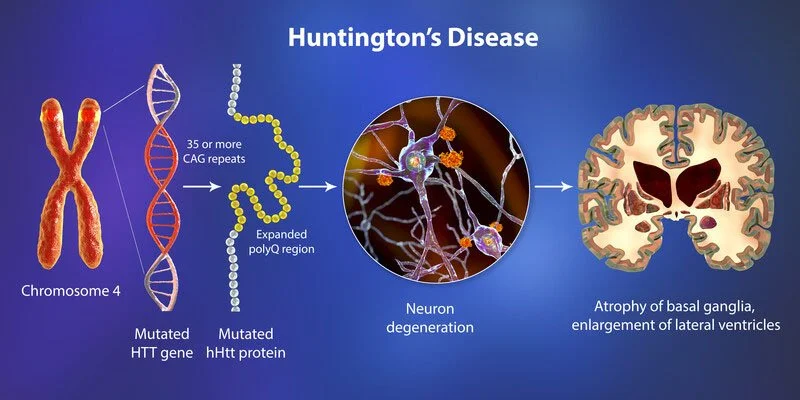
HD is caused by a mutation in the huntingtin (HTT) gene, which provides instructions for making a protein called huntingtin. The mutated huntingtin protein accumulates in the brain, leading to the death of nerve cells and the progressive loss of physical and mental abilities.
Symptoms of HD usually develop in midlife, although they can occur earlier or later. The most common symptoms include involuntary movements, difficulty with balance and coordination, problems with thinking and memory, and changes in mood and behavior.
There is no cure for HD, and treatment is primarily focused on managing the symptoms. Medications can be used to help control the involuntary movements and mood changes, while physical therapy and occupational therapy can help maintain mobility and independence. Genetic counseling and testing are also available for individuals with a family history of HD.
Huntington’s disease is rare. This disease has a wide impact on an individual’s functional abilities and commonly results in movement disorders, thinking (cognitive) disorders, and psychiatric disorders. Huntington’s disease is incurable. The early signs include coordination problems and memory lapses. In the later stages, people often require full nursing care.
This disease symptom could develop at some time, but they typically first appear when individuals are in their 30s or 40s. If the condition develops before age 20, it is named juvenile Huntington’s disease. When this disease develops early, the symptoms. are somewhat varied. and the disease might progress faster. Medications are available to assist manage the symptoms of Huntington’s disease.
What is Huntington’s?
Huntington’s disease is a neurological condition. This disease is an inherited disease that results from a gene mutation. Toxic proteins collect in the brain and lead to damage, leading to neurological symptoms. Huntington’s disease (HD) is unique among other neurodegenerative diseases.
This disease is caused by abnormal huntingtin protein. This type of disease is an inherited condition that leads to involuntary muscle movements and changes in cognition and behavior. Many individuals live for many years without symptoms. After symptom onset, individual live about 15 years to 18 years but might live longer than twenty five years.
As the disease affects various parts of the brain, it impacts cognition, movement, and behavior. It becomes harder to reason, walk, think, swallow, and talk. Eventually, the person will require full-time care. The condition or its complications could be fatal. There is currently no cure, but treatment could assist with symptoms. Huntington’s disease (HD) is an inherited disorder. this disease leads to nerve cells (neurons) in parts of the brain slowly breaking down and dying. The disease attacks areas of the brain that assist to hold voluntary (intentional) movement, as well as other areas. individuals living with Huntington’s disease develop uncontrollable dance-such as movements (chorea) and abnormal or not normal body postures, as well as problems with behavior, emotion, thinking, and personality.
For example, uncontrolled movements in the people’s fingers, feet, face, or torso. These movements are signs of chorea. They can get more intense when the person is nervous or distracted; as Huntington’s disease progresses, the person’s movements can become more extreme and obvious. Symptoms of Huntington’s disease typically appear in middle-aged people (adult Huntington’s disease). They can also appear in children (juvenile Huntington’s disease), but this is rare. The disease gets worse over time. Early signs of Huntington’s disease could vary but typically include mild clumsiness or problems with balance or movement, cognitive or psychiatric symptoms (problems with thinking or emotion), and changes in behavior.
For some individuals, chorea can make it harder to walk, which raises the chances of falling. A few individuals with this disease do not develop chorea; rather, they may become rigid (stiff) and move very little or not at all. This condition is called akinesia. Other people may start with chorea but become rigid as the disease progresses. In addition to chorea, a few individuals have unusual fixed (unchanging) postures, which is known as dystonia. The two movement disorders (dystonia and akinesia) can alternate or blend.
Other symptoms may include tremors (unintentional back-and-forth movement in a person’s muscles) and unusual eye movements. Eye movements can occur early in a disease. Physical changes might include slurred speech and problems with swallowing, eating, speaking, and particularly walking. Individuals with this disease might weight lose because of problems with feeding, swallowing, choking, and chest infections. Other symptoms might include insomnia (having trouble sleeping), energy loss, fatigue, and seizures. Ultimately, the person would require to stay in bed or a wheelchair. Changes in thinking (cognitive changes) may include problems with attention or judgment and difficulty solving problems or making decisions. Other changes might be including trouble with driving, prioritizing (deciding which things are more important to do and which are less important), difficulty or problem organizing, learning new things, remembering a fact, putting thoughts into words, or/and answering a question. These cognitive changes get worse as the disease progresses until people with Huntington’s disease are not able to work, drive, or care for themselves.
When the cognitive problems are severe enough that the individual cannot function in daily life, the condition is described as dementia. But many individuals with Huntington’s disease stay aware of their environment and can express their emotions. Changes in behavior might include mood swings; feeling irritable (cranky); not being active; or/and feeling apathetic (uninterested or not interested), depressed, or angry. These symptoms may reduce as the disease progresses. But in a few individuals, the symptoms could continue and might involve angry outbursts, thoughts of suicide, deep depression, and psychosis (losing touch with reality). Individuals with Huntington’s disease may withdraw from social activities.
Huntington’s disease has 2 subtypes:
- Adult-onset Huntington’s disease. Adult-onset Huntington’s disease is the most common form of this type of disease. the person typically develops the symptoms in their mid-30s and 40s.
- Early-onset Huntington’s disease. In rare instances, a child or adolescent will develop a disease. A Child with the disease tends to experience abrupt difficulties with schoolwork. and typically has symptoms that are quite the same as Parkinson’s disease.
Mechanism of Injury / Pathological Process
The most or more obvious neuropathology in this disease occurs within the neostriatum (Basal Ganglia part), comprising gross atrophy of the caudate nucleus and putamen, accompanied by selective neuronal loss and astrogliosis (an abnormal raise in the number of astrocytes). the marked or significant neuronal loss also is seen in a deep layer of the cerebral cortex. another or a few regions, involving a globus pallidus, the thalamus, the substantia nigra, the subthalamic nucleus, and the cerebellum, see differing degrees of atrophy depending on the stage of the disease.
Pathophysiology
In Huntington’s disease, there is an excessive or extreme sequence of the CAG repeats in part of the HTT (“Huntingtin”) gene, which is situated on the short arm of chromosome 4. Normal individuals have between nine CAG and thirty-five CAG repeats, while individuals diagnosed with this type of disease, as well as carriers, have an abnormal or not normal expansion accommodating 36 or more CAG repeats. The HTT gene, or Huntington’s disease gene, codes for a protein called huntingtin. This type of protein is found in the neurons throughout the brain, and its normal function is unknown. In an affected person, neuronal degeneration initiates in the striatum. and it progresses to the cerebral cortex, following the pattern that correlates to the clinical progression of this type of disease. The project first identified the mutation in the HTT gene as the lead to Huntington’s disease in 1993.
Inheritance of Huntington’s disease
The mutation is a dominant gene, so if a parent has Huntington’s disease then the children have a 50-50 risk of inheriting it. There is a genetic test to make or confirm a diagnosis of Huntington’s disease. Utilizing a sample of the blood, the genetic test analyses DNA( deoxyribonucleic acid ) for this type of disease mutation by counting the number of CAG repeats in the huntingtin gene. Individuals who do not have Huntington’s disease generally have 28 or fewer repeats. Individuals with Huntington’s disease usually have 40 or more repeats, a few times more than 100.
Who does Huntington’s disease (HD) affect?
However anyone can develop Huntington’s disease, it tends to run in individuals of European descent (having family members who came from Europe). But the main factor is whether people have a parent with Huntington’s disease. If people do, they have a 50% chance of also having the disease.
How is Huntington’s disease (HD) inherited?
To understand how this type of disease is inherited, individuals have to know a little bit about genetics — the study of physical characteristics passed down from generation to generation. Every cell in the body has DNA (deoxyribonucleic acid). DNA is the body’s instruction manual. It provides the information people need to repair and rebuild cells. DNA dictates everything from people’s hair color and height to how their organs function. Genes are such as “chapters” within the DNA instruction manual. Here’s how genes affect Huntington’s disease:
- The huntingtin gene (HTT or Huntington’s disease gene) tells the body how to build the huntingtin protein. people get one HTT gene from every parent.
- If an individual has this disease, one of the parents passed on an HTT gene with a mutation. It tells the body to create an unusually long protein. Researchers believe this long protein damages and kill brain cells.
- Someone who inherits the gene will eventually develop Huntington’s disease symptoms. The exact age when symptoms appear varies. Often, Huntington’s disease symptoms start earlier in every new generation than in the previous generation.
How does Huntington’s disease (HD) affect the brain?
The parts of the brain affected include the areas named the basal ganglia and cerebral cortex. These interconnected areas are associated with various types of activity including movement, learning, thinking, planning, motivation, and emotion.
This type of disease develops when misshapen proteins destroy the neurons (brain cells). First, they normally attack The basal ganglia, the area in the brain that oversees the body movements people control. A disease also impacts a brain’s cortex (surface of the brain). This part of the brain assists with thinking, decision-making, and memory.
What are the symptoms of Huntington’s disease?
It generally causes movement disorders, cognitive disorders, and psychiatric disorders with a wide or broad spectrum of signs and symptoms. Which symptoms appear first differs greatly from individual to individual. A few symptoms appear more dominant or/and have a more impact on functional ability, but those could alter throughout a disease or condition.
Signs and symptoms are most likely to appear in people aged 30–50 and can occur at any age. Key symptoms include:
- personality and mood changes
- depression
- problems with memory, thinking, and judgment
- loss of coordination and control of movements
- difficulty swallowing and speaking
The development of symptoms can vary between individuals. A few would experience depression first and then changes in their motor skills. unusual behavior and mood changes. These two are common early signs.
Early signs and symptoms
A doctor might not recognize or identify the early symptoms if none of the individual’s family members have previously received a diagnosis of this disease. It could take time to reach the diagnosis. Initial signs and symptoms may include:
- slight uncontrollable movements
- small changes in coordination and clumsiness
- stumbling
- slight mood and emotional changes
- difficulty focusing and functioning at work or school
- lapses in short-term memory
- depression
- irritability
The people might also lose motivation and focus, appearing lethargic and lacking initiative. Other possible early signs of this disease may include dropping things and forgetting people’s names. Although, most people do these things occasionally. Over time, these symptoms become more severe in people with Huntington’s disease.
The middle and later stages
As the disease progresses, it involves many physical changes, loss of motion control, emotional changes, and cognitive changes.
Physical changes
The people may experience:
- difficulty or problem speaking, including problems with finding words and slurring
- weight loss, leading to weakness
- difficulty eating and swallowing, as the muscles in the mouth and diaphragm might lose function
- risk of choking, specifically in the later stages
There may be uncontrollable body movements, including:
- movements of the face
- jerking of the parts of the face and the head
- flicking or fidgety the movements of the arms, legs, and body
- lurching and stumbling
Over time, uncontrollable movements happen more often and usually with more intensity. Ultimately, those might become slower as the muscles become more rigid.
Emotional changes
There are two types of categories of emotional symptoms and psychological symptoms of Huntington’s disease. The first category involves developing mental illnesses that are also common among individuals who do not have the disease. About 40% of individuals with this disease would develop conditions such as obsessive-compulsive disorder, mania, or delusional disorders. A second type includes the mental state changes that lead to brain alterations as a result of HD (Huntington’s disease) itself. These can include:
- aggression
- anger
- antisocial behavior
- apathy
- depression
- excitement
- frustration
- increasingly apparent lack of emotion
- moodiness
- stubbornness
- cognitive changes
There may also be:
- a loss of initiative
- a reduction of organizational skills
- disorientation
- difficulty focusing
- problems with multitasking
According to the disease Society of America (HDSA), the risk of suicide is elevated in individuals with Huntington’s by up to 10 times the national average due to cognitive changes, including disinhibition and impulsivity. It recommends that caregivers of individuals with the condition are aware of the signs of suicide ideation.
The later stage
Eventually, the people will no longer be able to walk or talk and will require full nursing care. though, they would typically understand most of what they hear and be aware of their surroundings, as well as their friends and family members.
Movement disorders
The movement disorders associated with HD (Huntington’s disease) could involve both involuntary movement problems and impairments in voluntary movements, like:
- Involuntary jerking or writhing movements (chorea)
- The muscle problems, such as rigidity or muscle contracture (dystonia)
- Slow or unusual eye movements
- Impaired gait, posture, and balance
- Difficulty with speech or swallowing
Impairments in voluntary movements — instead of involuntary movements — might have a greater impact or effect on an individual’s ability to work, perform daily activities or regular activities, communicate, and remain independent.
Chorea (derived from the Greek word meaning a dance) is the most common movement disorder seen in this type of disease. Initially, mild chorea may lead the patient to appear restless as if they are fidgeting. This progresses, and severe chorea might appear as uncontrollable or not controllable writhing and flailing of the extremities, which interferes with function. As the disease or condition progresses, the chorea coexists with and slowly is replaced by ataxia, dystonia, and parkinsonian characteristics, like bradykinesia (which means slowness of movement), rigidity, and postural instability. In advanced disease, persons develop the akinetic-rigid syndrome, with minimal or no chorea. and other late features such as spasticity, clonus, and extensor plantar responses. Dysarthria and dysphagia are common. Abnormal eye movements might be seen early in the disease. Other movement disorders, such as tics and myoclonus, might be seen in patients with Huntington’s disease.
The motor symptoms and signs
The characteristic motor changes are involuntary, unwanted movements. Initially, the movements frequently happen in the distal extremities such as the fingers and toes, but also in the small facial muscles. For bystanders, these muscle twitches are usually invisible or can be explained as nervousness. In daily life, walking becomes unstable and the person could look as if people are slightly drunk. Gradually unwanted movements or activities spread to other muscles from the distal to more proximal and axial. The choreatic movements are present all the period the patient is awake or wakeful. No single pattern exists, but facial choreatic movements could lead to the continuous movement or motion of the facial muscles where for instance an eyebrow is lifted, a closed eye, and the head is bent or/and turned while the tongue is protruded with the lips pouting. The most prominent is the extension movements of the long back muscles. Talking and swallowing slowly become more problematic leading to choking at any time in a few patients. In the later stages, the patient even becomes mute.
Dysarthria and dysphagia become very prominent during a disease. patients develop hypokinesia, akinesia, and rigidity leading to a slower pace of all activities like bradykinesia (it means slowness of movement) and severe or more hesitation in embarking on a movement (akinesia: difficulty in starting movements)). A balance between the chorea and the hypokinesia is determined separately or particularly. The extremes are on the one hand or single hand the youthful patient or person with an overwhelming rigidity (Westphal variant). on another hand, the very old patient is severely or more affected in the last stage of the disease or condition with a long duration of illness, and bed-bound with rigidity. and flexion contractures in the extremities.
Dystonia is characterized by slower movements with a raised muscle tone leading to abnormal posture, for instance, torticollis, but also rotation of the trunk or limbs. Dystonia (for instance torticollis) could be the first motor sign of Huntington’s disease. another unwanted movement includes or involves tics, comparable or equivalent to the ones seen in Tourette syndrome, but these are fairly rare. Cerebellar signs could appear sporadically, the same as the presence of hypo- and hypermetria. Walking is usually described as ‘drunk’ or ‘cerebellar ataxia’-like. Distinguishing between ataxic walking and choreatic walking is very difficult or problematic. Pyramidal signs (Babinski sign) are present incidentally. The influence of motor disturbance on everyday activities of life progresses over time.
The presence of hyperkinesia and hypokinesia outcome in problems in walking and standing. and often reasons for an ataxic gait and repeated falls. Furthermore, activities in daily life such as getting out of bed, taking a shower, dressing, toileting, cleaning the house, cooking, and eating become more and more difficult. Depending on the kind of work a patient does, motor signs would sooner or later interfere with performance, even if psychiatric and cognitive changes are still in the background.
Cognitive disorders
Cognitive impairments frequently associated with Huntington’s disease include:
- Difficulty organizing, prioritizing, or focusing on tasks
- Deficiency of flexibility.
- lack of a tendency to get stuck on a thought, behavior, or/and action (perseveration)
- Lack of or Deficiency in impulse control that could be resulting in outbursts, acting without thinking, and sexual promiscuity
- Lack of awareness of one’s behaviors and an abilities
- Slowness in processing thoughts or ”finding” words
- Difficulty in learning new information
Cognitive decline is a feature of Huntington’s disease, but the rate of progression among particular patients could vary considerably. Dementia and the psychiatric features of Huntington’s disease are often the earliest symptoms. Slowing of cognition, impairment of intellectual function, and memory disturbances usually happen later.
Psychiatric disorders
The most common psychiatric disorder associated with this disease is depression. This is not simply the reaction to receiving a diagnosis of this disease. Instead, depression appears to happen because of injury to the brain and the subsequent changes in brain function. Signs and symptoms may include:
- Feelings of irritability, sadness, or apathy
- Social withdrawal
- Insomnia
- Fatigue and loss of energy
- Frequent thoughts of death, dying, or suicide
Other common psychiatric disorders include:
- Obsessive-compulsive disorder. this is a condition marked by recurrent, intrusive thoughts and repetitive behaviors.
- Mania. this can lead to elevated mood, overactivity, impulsive behavior, and inflated self-esteem.
- Bipolar disorder. this is a condition with alternating episodes of depression and mania.
Behavior and psychiatric symptoms and signs
Psychiatric symptoms are very often present in the early stage of the disease, often before the onset of motor symptoms. The percentage of patients with psychiatric signs differs between 33% and 76% depending on the methodology of the study. Because of their impact on daily life, these symptoms and signs typically have a highly negative impact on functioning and the family. The most frequently happening sign is depression. A diagnosis is difficult because weight loss, apathy, and inactivity or not activity also occur in this disease. Generally, there is low self-esteem, feelings of guilt, and anxiety. Apathy is related to the stage of disease, whereas anxiety and depression are not. Suicide happens more commonly in earlier symptomatic individuals and also in premanifest gene carriers. Around the time of the gene test and the stage when independence reduces are the riskiest periods for suicide.
Anxiety also occurs frequently (34-61%), sometimes about uncertainty about the start and or the course of the disease. The compulsions and obsessions could disturb a patient’s life and also lead to irritability and aggression. Irritability is often the very first sign, in retrospect, but in fact, happens during all stages of the disease. The way irritability is expressed differs enormously from serious disputes to physical aggression. A loss of interest and raising passive behavior are seen as part of the apathy syndrome. It could be difficult to discriminate apathy from depression. Psychosis might appear, mainly in a later stage of the disease. In most cases, these go together with cognitive decline. A complete clinical image is comparable to schizophrenia with paranoid and acoustic hallucinations. In the early stages, hyper-sexuality can lead to considerable problems in a relationship. In the later stages, hypo-sexuality is a rule.
Dementia
Cognitive decline is the other main sign of Huntington’s disease and can be present long before the first motor symptoms appear, but can also be very mild in far advanced stages of the disease. The cognitive changes specifically concern executive functions. In normal conditions, cognitive and motor behavior is goal-directed and planned. Normally individuals could distinguish what is relevant and what can be ignored, but patients with Huntington’s disease lose this capability. The patients are no longer able to organize their life or to plan things that in the past were simple. They lose the flexibility of mind, and could no longer make mental adjustments. The misjudgments lead to complicated situations, with patients no longer reacting as they did in a past or/and in a way that the environment expects. Language is relatively spared. Memory certainly becomes impaired, though semantic memory can be spared to a certain extent. All psychomotor processes become severely retarded.
What is juvenile Huntington’s disease (HD)?
If symptoms begin before a person is 20, the doctor will diagnose them with juvenile Huntington’s disease. The physical symptoms of the juvenile version of the disease tend to be various and can include leg stiffness, tremors, and regression in learning. This version of the disease generally progresses more rapidly. It might be fatal within 10 years of a diagnosis. The lead to death is usually a complication, such as pneumonia or choking.
Symptoms of juvenile Huntington’s disease
The beginning and progression of this (Huntington’s disease) disease in young individuals might be slightly different from those in adults. Problems that frequently present early in the course of the disease include:
Behavioral changes
- Difficulty paying attention
- The rapid, considerable drop in overall school performance
- Behavioral problems
Physical changes
- Contracted and rigid muscles that affect gait (specifically in young children)
- Tremors or slight involuntary movements
- Frequent falls or clumsiness
- Seizures
What are the causes of Huntington’s disease?
This disease is caused by the inherited difference in a single gene. Huntington’s disease is an autosomal dominant disease. which means that an individual requires only one copy of the nontypical gene to develop the disorder.
Except for genes on the sex chromosomes, an individual inherits two copies of every gene — one copy from each parent. The parents with a nontypical gene could pass along the nontypical copy of the gene or a healthy (or normal) copy. Each child in a family, thus, has a 50% chance of inheriting the gene that reason to the genetic disorder.
Huntington’s disease is caused by a mutation in a gene for the protein called huntingtin. Most people have fewer than 27 CAG repeats in their Huntington’s disease gene, so they are not at risk for the disease.
What are the complications of Huntington’s disease?
After Huntington’s disease starts, a people’s functional abilities gradually worsen over time. The rate of disease duration and progression differs. The time from the first symptoms to death is frequently about 10 years to 30 years. Juvenile Huntington’s disease commonly outcomes in death within ten years after symptoms develop.
The clinical depression associated with Huntington’s disease may raise the risk of suicide. A few research suggests that a more risk of suicide happens before a diagnosis is made and in the middle stages of the disease when an individual begins to lose independence.
Pneumonia and heart disease are the two leading causes of death for individuals with Huntington’s disease. In Additional, this type of disease patients has a higher incidence of choking and respiratory complications, gastrointestinal diseases (like cancer of the pancreas), and suicide than the non-Huntington’s disease population.
Ultimately, a person with Huntington’s disease needs help with all activities of daily living and care. Late in the disease, the individual would likely be confined to a bed and unable to speak. The individual with this (Huntington’s disease) disease is typically able to understand language and has an awareness of family and friends, though a few won’t recognize family members. Common causes of death include:
- Pneumonia or other infections
- Injuries related to falls
- Complications related to the inability to swallow
What is the prevention of Huntington’s disease?
An individual with a known the family history of this disease is understandably concerned about whether those might pass the Huntington’s gene on to those children. These individuals might consider genetic testing and family planning options.
If an at-risk parent is considering genetic testing, it could be helpful to meet with a genetic counselor. A genetic counselor would discuss the potential risks of a positive test result, which would indicate that the parent would develop a disease. Also, couples would require to make additional or extra choices about whether to have a child or to consider alternatives, such as prenatal testing for the gene or in vitro fertilization with donor sperm or eggs.
Other options for couples are in vitro fertilization and preimplantation genetic diagnosis. In this type of process, eggs are removed from an ovary and fertilized with the father’s sperm in a laboratory.
Can Huntington’s disease (HD) cause dementia?
In its later stages, Huntington’s disease can lead to dementia. The loss of brain function leads to personality changes and memory loss. Earlier in the disease, the impact on the brain is different. people might have problems multitasking or doing something that involves multiple steps. that could also be difficult to plan. or/and problem-solve soon after the outset of HD (Huntington’s disease).
Differential Diagnosis
When chorea is the presenting and more prominent sign. it taking history is the first and most valuable step. The frequently happening differential diagnoses for motor sign chorea. In many cases, the underlying lead is another general internal disorder or an iatrogenic disorder. Only very few genetically determined disorder is responsible for choreatic syndromes. In about 1% of the cases clinically diagnosed as Huntington’s disease by a clinician, the genetic test does not confirm the diagnosis. These are the so-named phenocopies. Phenocopies are defined by a clinical diagnosis of Huntington’s disease with chorea, psychiatric, and or cognitive signs and an autosomal dominant pattern of inheritance or the family history. In the last few years, many have been described
- Chorea Gravidarum
- Multiple Sclerosis
- Systemic Lupus Erythematosus (SLE)
- Neuroacanthocytosis
What is the diagnosis of Huntington’s disease?
A primary diagnosis of this disease is based primarily on the answers to questions, a general physical exam, a review of the family’s medical history, and neurological and psychiatric examinations.
Diagnosing Huntington’s Disease
To make the diagnosis of Huntington’s disease (HD), a genetic test, using a blood sample, will be performed. This type of test is generally combined with a complete medical history and other neurological and laboratory tests. For individuals who are at risk of carrying the Huntington’s disease gene, testing can be performed before symptoms occur.
Family history plays a major or main role in a diagnosis. the doctor will ask people questions about their medical background and give them a physical exam. If an individual and the doctor suspect Huntington’s disease, a neurologist would conduct more tests. A neurologist may test:
- Reflexes
- Muscle strength
- Balance
- Sense of touch
- Vision
- Hearing
- Mood and mental status
- Memory
- Reasoning
- Thinking skills
Neurological examination and medical history
The neurologist will conduct an in-depth interview to obtain the medical history (including any family history, named a pedigree or genealogy) to rule out other conditions. Neurological and physical exams might review the reflexes, balance, movement, muscle tone, hearing, walking, and mental status. Laboratory tests might also be ordered, and individuals with Huntington’s disease might be guided to specialists such as psychiatrists, genetic counselors, clinical neuropsychologists, or speech pathologists for specialized management and/or diagnostic clarification.
The neurologist will ask people questions and conduct relatively simple tests of the:
- Motor symptoms, such as reflexes, the strength of the muscle, and balance.
- Sensory symptoms include a sense of vision, touch, and hearing.
- Psychiatric symptoms, such as mental status and mood.
Neuropsychological testing
The neurologist might also perform standardized tests to check people:
- Memory
- Reasoning
- Mental agility
- Language skills
- Spatial reasoning
Psychiatric evaluation
People will likely be referred to a psychiatrist for an examination to look for several factors that could contribute to their diagnosis, including:
- Emotional state
- Patterns of behaviors
- Quality of judgment
- Coping skills
- Signs of disordered thinking
- Evidence of substance abuse
Diagnostic imaging
In a few cases, particularly if an individual’s family history and genetic testing. are inconclusive, the physician might recommend brain imaging, like computed tomography (CT) or, more likely, magnetic resonance imaging (MRI). As the disease progresses, these scans generally reveal shrinkage in parts of the brain and enlargement of fluid-filled cavities within the brain called ventricles. These changes do not necessarily indicate Huntington’s disease, because they can happen in other disorders. A person can have early symptoms of Huntington’s disease and still have normal findings on a structural computed tomography or magnetic resonance imaging scan.
Brain-imaging and function tests
The provider might order brain-imaging tests for assessing the structure of the brain or/and function of the brain. The imaging technologies may include magnetic resonance imaging scans or computed tomography scans that show detailed images of the brain. These pictures might reveal changes in the brain in areas affected by this disease. These modifications might not show up early in the course of the condition. These tests could also be utilized to rule out other conditions that may be leading to symptoms.
Genetic counseling and testing
If symptoms strongly suggest Huntington’s disease. the provider might recommend a genetic test. for the nontypical gene. This test can confirm a diagnosis. It may also be valuable if there is no known family history of Huntington’s disease or if no other family member’s diagnosis was confirmed with a genetic test. But the test won’t provide information that might assist determine a treatment plan. Before having such a test, the genetic counselor would explain the benefits and drawbacks of learning test results. The genetic counselor could also question and answer about an inheritance pattern of this (Huntington’s disease) disease.
Genetic testing could ensure. or/and rule out a suspected genetic condition or assist identify an individual’s chance of developing or passing on a genetic disorder. Genetic testing makes it possible to predict with a higher degree of sure if a few would develop Huntington’s disease.
- The most effective and accurate procedure of testing for Huntington’s disease. it is named the direct genetic test. counts the number of CAG repeats in the Huntington’s disease gene, using DNA taken from a blood sample. The presence of 36 or more repeats supports the diagnosis of Huntington’s disease. A test result of twenty-six or fewer repeats rules out Huntington’s disease.
- An older genetic test, named linkage testing (also called prenatal exclusion testing) needs a sample of DNA from a closely related affected relative, preferably a parent, to identify markers close to the Huntington’s disease gene and to determine if a fetus has inherited a chromosome 4 mutation from an affected grandparent. A version of the linkage method is a few times used for prenatal testing.
- Prenatal testing is an option for people who have a family history of Huntington’s disease and are concerned about passing the disease to a child. The prenatal testing could be done using either the direct procedure or the linkage method. As with adult testing, a direct method provides higher certainty.
Predictive genetic test
A genetic test can be given if people have a family history of the disease but do not have symptoms. This is called predictive testing. The test can not tell people when the disease will begin or what symptoms will appear first. A few individuals might have the test because they found not knowing to be more stressful. Others might want to take the test before having a child. The risks might include problems with insurability or future employment and the stresses of facing the fatal disorder. In principle, federal laws exist that make it illegal or not legal to use genetic testing information or details to discriminate against individuals with genetic diseases. These types of tests are only performed after consulting with the genetic counselor.
What is the treatment for Huntington’s disease?
No treatment can stop or reverse Huntington’s disease, but a few of the symptoms can be treated:
- The drugs tetrabenazine and deutetrabenazine could treat chorea associated with Huntington’s disease
- The antipsychotic drugs might relieve chorea and assist to control hallucinations, delusions, and violent outbursts
- Drugs might be prescribed to treat depression and anxiety
The side effects of drugs utilized to treat the symptoms of this (Huntington’s disease) disease. might involve fatigue, sedation, diminished concentration, restlessness, or hyperexcitability. These drugs should be only used when Huntington’s disease symptoms create problems for the individual living with Huntington’s disease.
No treatments could alter the course of Huntington’s disease. But medications could lessen a few symptoms of movement and psychiatric disorders. And numerous interventions could assist an individual to adapt to changes in abilities for a certain amount of time.
Medications will likely evolve throughout the disease, depending on overall treatment goals. Also, drugs that treat a few symptoms may result in side effects that worsen other symptoms. Treatment goals would be regularly reviewed and updated.
Options include:
- physiotherapy, occupational therapy, and speech therapy.
- medication to alleviate mood changes.
- medication to assist with jerky movements and difficulty swallowing.
- financial support through Centrelink.
- respite care, at the home or in residential facilities.
- supported accommodation including private residential facilities. shared supported accommodation. accommodation for young individuals with this disease, and residential aged care.
Medications for movement disorders
The drugs to treat movement disorders include the following:
- The drugs control movement. includes tetrabenazine (Xenazine) and deutetrabenazine (Austedo), which have been particularly approved by the Food and Drug Administration to suppress the involuntary jerking and writhing movements (chorea) associated with Huntington’s disease. These drugs don’t have any effect on the progression of the disease, although. Possible side effects include drowsiness, restlessness, and the risk of worsening or triggering depression or another psychiatric condition.
- Antipsychotic drugs, like haloperidol and fluphenazine, have the side effect of suppressing movements. Therefore, they might be beneficial in treating chorea. although, these drugs might worsen involuntary contractions (dystonia), restlessness, and drowsiness.
- Other medications. this medication such as olanzapine (Zyprexa) and aripiprazole (Abilify), might have fewer side effects but still should be utilized with caution, as they might also worsen symptoms.
- Another medication that might assist suppress chorea. involves this medicine such as amantadine (Gocovri, Osmolex ER), levetiracetam (Keppra, Elepsia XR, Spritam), and clonazepam (Klonopin). Though, side effects might limit their use.
Medications for psychiatric disorders
The Medications to treat psychiatric disorders would vary depending on the disorders and symptoms. Possible treatments include the following:
- Antidepressant drugs include such drugs as citalopram (Celexa), escitalopram (Lexapro), fluoxetine (Prozac), and sertraline (Zoloft). These drugs might also have some effect on treating obsessive-compulsive disorder. Side effects might include nausea, diarrhea, drowsiness, and low blood pressure.
- Antipsychotic medication such as quetiapine (Seroquel) and olanzapine (Zyprexa) might suppress violent outbursts, agitation, and another symptom of mood disorders or psychosis. Although, these drugs may cause different movement disorders themselves.
- Mood-stabilizing drugs that can assist prevent the highs and lows associated with bipolar disorder include anticonvulsants, such as divalproex (Depakote), carbamazepine (Tegretol, Carbatrol, Epitol, others) and lamotrigine (Lamictal).
Psychotherapy
A psychotherapist — a psychiatrist, psychologist, or clinical social worker — can provide talk therapy to assist with behavioral problems, develop coping strategies, manage expectations during the progression of the disease, and assist family members to communicate with each other.
Speech therapy
Huntington’s disease could particularly impair the control of muscles of the mouth and throat that are essential for speech, eating, and swallowing. A speech therapist can assist improve people’s ability to speak clearly or teach them to use communication devices — such as a board covered with pictures of everyday items and activities. Speech therapists could also address difficulties with muscles used in eating and swallowing.
Physical therapy
A physical therapist can teach people appropriate and safe exercises that improve strength, flexibility, balance, and coordination. These exercises can assist maintain mobility as long as possible and may lower the risk of falls. Instruction on appropriate posture and the use of supports to enhance posture may assist lessen the severity of some movement problems. When the use of a walker or wheelchair is needed, the physical therapist can provide instruction on the appropriate use of the device and posture. Also, exercise regimens could be adapted to suit the new level of mobility. A physiotherapist can assist with mobility and balance. It uses a range of treatments, including manipulation, massage therapy, exercise, electrotherapy, and hydrotherapy. people may be referred to a physiotherapist through their GP or social services.
A physiotherapist working with these clients should take note of a recent finding.
- People with Huntington’s disease need extra time to carry out everyday tasks.
- People with Huntington’s disease are forgetful but do not have classical amnesia. Those have the potential to benefit from memory aids.
- A few tasks, like walking and talking, which under normal circumstances might be regarded as relatively “automatic”, need more conscious attention in people with Huntington’s disease. They are more demanding of attentional resources. Individuals with Huntington’s disease require to focus on one activity at a time.
- Individuals with Huntington’s disease may not initiate activities but with encouragement could engage successfully in them and experience enjoyment.
Standard physiotherapy for Huntington’s disease includes:
- Gait re-education
- Balance retraining
- Fall prevention/management
- Aerobic capacity
- Muscle strengthening
- Wheelchair prescription and training
- Respiratory function
- Task-specific reach, grasp, and manipulation
Occupational therapy
An occupational therapist could assist the individual with this (Huntington’s disease) disease. and family members, and caregivers with the use of assistive devices that enhance functional abilities. These strategies may include:
- Handrails at home.
- The assistive devices for activities like bathing and dressing.
- Eating and drinking utensils are adapted for individuals with limited fine motor skills.
What are the Lifestyle and home remedies for Huntington’s disease?
Managing Huntington’s disease affects individuals with the disorder. family members, and other in-home caregivers. As the disease progresses, people will become more dependent on caregivers. many issues would need to be addressed, and the ways to cope with them will change over time.
For now, treating Huntington’s involves managing symptoms:
- Medications can help control fidgety movements. the doctor can work closely with people to manage any side effects and to change medications if needed.
- a few antipsychotic drugs have side effects that control movement and have been helpful for a few people.
- Antidepressants can also assist with obsessive-compulsive disorder.
- Mood-stabilizing drugs can reduce symptoms of mood disorders but may cause other side effects.
- Speech therapy or language therapy may be helpful for any problems with speech or swallowing.
- Occupational or physical therapy may help people learn how to better control movements. And assistive devices like handrails could assist individuals to manage those changing physical abilities.
- The nutritional support ranges from utilizing particular utensils to concentrating on nutrient-dense foods to supplementing with tube feeding in the later stages.
- Exercise may be very helpful. individuals with Huntington’s people stay as fit and active as they could seem to do good than those who do not.
- Psychotherapy can teach people ways to manage changes in their emotions and how they think.
- The strategies like breaking tasks into easy steps might go a long way toward making these changes a bit easy for individuals and their families.
Family members could assist by making a few changes at the home:
- Serve extra meals and add high-calorie supplements to help people stay at a healthy weight.
- Limit distractions during meal times.
- Choose foods that are easier to swallow and chew.
- Use forks and other utensils made for individuals with limited motor skills.
- Use covered cups with straws or/and drinking spouts.
- Keep a routine.
- Use phone or computer reminders for tasks.
- Maintain life as calm, simple, and low-stress as possible.
- For a child, work with the school counselor to make an education plan.
- See friends and keep social interactions as much as possible.
- Add the wheelchair ramps
- Add elevators to the home if possible.
- Add safety bars in the bathrooms, next to the bed, and on the stairs.
- Use voice-controlled lights and another “smart” home feature.
- Use electronic speech programs or/and picture charts to aid communication.
Eating and nutrition
Factors regarding nutrition and eating include the following:
- Difficulty maintaining healthy body weight. This might be led to problems in eating, higher caloric requirements due to physical exertion, or unknown metabolic problems. To get adequate nutrition, people may require to eat more than three meals a day or use dietary supplements.
- Difficulty with chewing, swallowing, and fine motor skills. These problems can limit the amount of food people eat and raise the risk of choking. It may assist to remove distractions during a meal and select foods that are easier to eat. Utensils are designed for individuals with limited fine motor skills. and covered cups with straws or drinking spouts also could assist.
Ultimately, a person with Huntington’s disease will require assistance with eating and drinking.
Managing cognitive and psychiatric disorders
The relatives and caregivers could assist create an environment. and that might assist individuals with this (Huntington’s disease) disease, evade stressors and manage cognitive and behavioral challenges. These strategies include:
- Using calendars and schedules to assist keep a routine
- Initiating tasks with reminders or assistance
- Prioritizing or organizing work or activities
- Breaking down tasks into manageable steps
- Creating an environment that is as simple, calm, and structured as possible
- Identifying and avoiding stressors that could trigger outbursts, irritability, depression, or other problems
- For school-age a child or adolescents, consulting with school staff to develop an appropriate individual education plan
- Providing opportunities for the individual to maintain social interactions and friendships as much as possible
Coping and support
Several strategies might assist individuals with Huntington’s disease and their families cope with the challenges of the disease.
Support services
Support services for individuals with Huntington’s disease and families include the following:
- Nonprofit agencies, like this type of disease Society of America, provide caregiver education, referrals to outside services, and support groups for individuals with a disease and caregivers.
- Local and state health or social service agencies might provide daytime care for individuals with the disease, meal assistance programs, or respite for caregivers.
Planning for residential and end-of-life care
Because Huntington’s disease leads to the progressive loss of function and death. it is important to anticipate care that would be required in the advanced stages of the disease and near the end of life. The early debate about this type of care did not able an individual with this (Huntington’s disease) disease to be engaged in these decisions and to share what they want from their care. Creating legal documents that define end-of-life care could be beneficial to everyone. Those empower the individual with the disease, and they might assist family members to evade struggle late in the disease’s progression. The provider could offer advice on the advantage and drawbacks of care options at a time when all choices could be carefully considered. Matters that may require to be addressed include:
- Care facilities. Care in the advanced stages of the disease will likely need in-home nursing care or care in an assisted living facility or nursing home.
- Hospice care. Hospice services provide care at the end of life that assists a person approach death with as little discomfort as possible. This care also provides support and education to family members to assist them to understand the process of dying.
- Living wills. Living wills are legal documents that enable people to spell out care preferences when it is not possible to make decisions. For eg, these directions might indicate whether. or not the individuals want life-sustaining interventions. or aggressive treatment of an infection.
- Advance directives. These legal documents enable people to identify one or more people to make decisions on their behalf. people may create an advance directive for medical decisions or financial matters.
When to see a doctor
See the healthcare provider if individuals notice modifications in their movements, emotional state, or/and mental ability. The signs and symptoms of Huntington’s disease could be caused by several different conditions. so, that is significant to get a prompt, thorough diagnosis.
Preparing for the appointment
If individuals have a few of the signs or symptoms associated with this (Huntington’s disease) disease. they would like to be suggested to a neurologist doctor after an initial visit to the provider. A review of the symptoms, mental state, medical history, and family medical history could all be important in the clinical assessment of a potential neurological disease.
What people can do
Before the appointment, make a list that includes the following:
- Signs or symptoms — or any changes from what is normal for people— that may be leading to concern
- Recent changes or stresses in the life
- All medications — including drugs available without a prescription and dietary supplements — and doses people take
- Family history of Huntington’s disease or other disorders that may lead to movement disorders or psychiatric conditions
People might want a family member or friend to accompany them to their appointment. This individual could provide support and offer various perspectives on the impact of symptoms on functional abilities.
What to expect from the doctor?
The provider is likely to ask people several questions, including the following:
- When did people begin experiencing symptoms?
- Have the symptoms been continuous or intermittent?
- Has anyone in the family ever been diagnosed with Huntington’s disease?
- Has anyone in the family been diagnosed with another movement disorder or/and psychiatric disorder?
- Are people having trouble performing work, schoolwork, or daily tasks?
- Has anyone in the family died young?
- Is anyone in the family in a nursing home?
- Is anyone in the family fidgety or moving all the time?
- Have people noticed a change in the general mood?
- Do people feel sad all of the time?
PROGNOSIS
How does Huntington’s disease (HD) typically progress?
Huntington’s disease can begin at different ages in different individuals. It gets worse over time.
Early stage
The symptoms are easier to handle earlier in the disease. the individuals might feel moody or/and clumsy and struggle with complex thinking. People may also have small uncontrollable movements, but typically, people can continue their everyday activities.
Middle stage
The Physical changes and mental changes. during the middle stage make working, driving and household upkeep not possible. people may begin to have trouble with swallowing, and people might lose weight. people’s balance may be off, increasing the risk of falling. People can still manage their care. Typically, people can handle bathing, getting dressed, and eating on their own or with some help.
End-stage
During the final stage of Huntington’s disease, people will need help with everything. people are usually unable to leave the bed. This is when most individuals receive care day and night.
DISCLAIMER:
This article is intended or purposeful for your general informational purposes only and does not address particular circumstances. it is not a substitute for professional advice(or guidance) or help( or assistance) and should not be relied on to make decisions of any kind. A few or any actions you take upon the information presented in this article are strictly at your own risk and responsibility.
FAQs
What are the 3 symptoms of Huntington’s disease?
The first symptoms of Huntington’s disease frequently include:
difficulty concentrating.
memory lapses.
depression – including a low mood, a lack of interest in things, and feelings of hopelessness.
stumbling and clumsiness.
the mood swings, like irritability or/and aggressive behavior.
What causes Huntington’s disease?
Huntington’s disease is caused by a mutation in a gene for the protein called huntingtin. Most people have fewer than 27 CAG repeats in their Huntington’s disease gene, so they are not at risk for the disease.
How long can a person live with Huntington’s disease?
This disease is a condition that stops the parts of the brain from working properly over time. It is passed on (inherited) from the person’s parents. It gets gradually worse over time and is generally fatal after a period of up to 20 years.
Is Huntington’s disease curable?
There is currently no cure for this disease or any way to stop it from getting worse. But treatment and support can help ease some of the problems caused by the condition.
What are the 5 stages of Huntington’s?
What Are The 5 Stages Of Huntington’s Disease?
Stage 1: Preclinical Stage.
Stage 1: Early Stage.
Stage 2: Early Intermediate Stage.
Stage 3: Late Intermediate Stage.
Stage 4: Early Advanced Stage.
Stage 5: Advanced Stage.
Treating Someone With Huntington’s Disease.
What are the 4 main symptoms of Huntington’s disease?
Symptoms
Involuntary jerking or writhing movements (chorea)
The muscle problems, such as rigidity or muscle contracture (dystonia)
Slow or unusual eye movements.
Impaired gait, posture, and balance.
Difficulty with speech or swallowing.
Who suffers from Huntington’s disease?
Frequency. Huntington’s disease affects an estimated 3 to 7 of every 100,000 individuals of European ancestry. The disease appears to be not more common in a few other populations. including individuals of Japanese, Chinese, and African descent.
How does Huntington’s disease cause death?
This type of disease(Huntington’s disease) is a progressive neurodegenerative autosomal dominant disease. those are described by choreatic and hypokinetic movements, disturbed behavior, and cognitive decline. Pneumonia is the most common lead to death, followed by cardiovascular diseases.
How can Huntington’s be treated?
No treatments could alter the course of Huntington’s disease. But medications can lessen a few symptoms of movement and psychiatric disorders. And multiple interventions could assist an individual to adapt to changes in abilities for a certain amount of time.
At what age does Huntington’s disease appear?
People can begin to show the symptoms of Huntington’s disease at almost any age. Most would develop problems between the ages of 30 years and 50 years. The condition slowly gets worse for around 10-25 years, until the individual dies.
Can you live a normal life with Huntington’s?
This disease makes each day’s activity more difficult to do over time. How fast it progresses differs from person to person. But the average lifespan after diagnosis is ten years to thirty years. Huntington’s disease itself is not fatal.
What is end-stage Huntington’s disease?
Advanced stages of Huntington’s
At this stage, individuals with this disease are no longer able to work. or/and manage those finances, individual care, and domestic responsibilities. and would have difficulty with mobility, and need to be in a bed or chair for more of the duration.
How is Huntington’s diagnosed?
To make the diagnosis of Huntington’s disease (HD), a genetic test, using a blood sample, will be performed. This type of test is generally combined with a complete medical history and other neurological and laboratory tests. For individuals who are at risk of carrying the Huntington’s disease gene, testing can be performed before symptoms occur.
Is Huntington’s disease fatal?
This disease (Huntington’s disease) is a fatal genetic disorder. that leads to a progressive breakdown of the nerve cells in the brain. Those deteriorate an individual’s physical and mental abilities. and typically during their prime working years. and have no cure.
How is Huntington’s spread?
This type of disease is autosomal dominant. and meaning the inheritance of just a single copy of the not normal chromosome from the biological parent is enough to lead to it. If one parent carries the abnormal or not normal gene, per of their biological children has a 50 percent chance of this disease inheritance.
How can people prevent Huntington’s disease?
Because Huntington’s is a genetic disease, individuals could not do anything to prevent it if people have inherited it. If individuals have a history of this disease in those families, they might wish to have genetic counseling before having children of their own.
Does Huntington’s disease come from Mom or Dad?
There have been reports that juvenile start Huntington’s chorea is almost always inherited from a father, and that late-onset Huntington’s chorea is inherited more frequently from the mother than from the father.
What are 3 interesting facts about Huntington’s disease?
Some facts about Huntington’s:
People can live with the faulty gene for years without symptoms, but if people do have it, at some stage people will develop symptoms. Doctors cannot tell people when this will be. Huntington’s disease affects men and women. It generally develops between the ages of 30 and 50 but can start at any age.
What is the average age of death for a few ones with Huntington’s?
Huntington’s disease is an inherited condition that leads to involuntary muscle movements and changes in cognition and behavior. Many individuals live for many years without symptoms. When symptoms started, most people live about 15 years to 20 years However they may vary, and may live longer than 25 years.
What kills Huntington’s?
People with Huntington’s disease generally die within 15 to 20 years of their diagnosis. The most common reasons for death are infections (such as pneumonia) and injuries related to falls.
What vitamins help with Huntington’s?
Mostly Doctors prescribe biotin and thiamine vitamin supplementation and it is also an approved treatment for BTBGD. and has been utilized with success for individuals with this type of condition(Huntington’s disease).
What food is not allowed for Huntington’s disease?
The problem the foods to swallow like flaky puff pastry, raw or uncooked vegetables, apples, pears, and high-fiber white bread should be evaded. Eating small but frequent amounts of high-calorie, nutrient-dense foods such as avocados, smooth nut butter, and hearty soups could prevent choking while ensuring adequate calorie intake.
What is the best diet for Huntington’s?
Choose soft, easy-to-chew, and easy-to-swallow foods (aim for the relevance of porridge). Use plenty of sauces and gravies to assist make main meals easier to swallow. Add plenty of ice cream, custards, and cream to desserts. Avoid or Evade hard foods such as nuts and lollies.
What is the first-line treatment for Huntington’s?
The medicines to control the movement include tetrabenazine (Xenazine) and deutetrabenazine (Austedo), which have been particularly approved by the Food and the Drug Administration to suppress the involuntary jerking and writhing movements (chorea) associated with Huntington’s disease.
What protein causes Huntington’s?
Huntington’s disease (HD) is various from other neurodegenerative diseases. in that, it is always inherited. This disease is caused by abnormal huntingtin protein.
What part of the brain does Huntington’s disease(HD) affect?
The parts of the brain affected include the areas named the basal ganglia and cerebral cortex. These interconnected areas are associated with various types of activity including movement, learning, thinking, planning, motivation, and emotion.
What treatment has the best utility for Huntington’s disease(HD)?
These medicines (haloperidol, tetrabenazine, and amantadine) could assist in controlling the unusual movements that led to HD (Huntington’s disease). Haloperidol and tetrabenazine can also assist prevent hallucinations and delusional thoughts. Depression and suicide are common among individuals with this disease.
What can a neurologist do for Huntington’s disease?
Because the nervous system is complex, a neurologist might specialize in a particular area, such as movement disorders. Movement disorders are associated with changes in brain cells. and that assists us to move. They would assess, manage, and treat; they are the “team leader”.
What are the most common complications of Huntington’s disease?
Pneumonia and heart disease are the two leading causes of death for individuals with Huntington’s disease. In Additional, this type of disease patients has a higher incidence of choking and respiratory complications, gastrointestinal diseases (like cancer of the pancreas), and suicide than the non-Huntington’s disease population.
2 Comments