
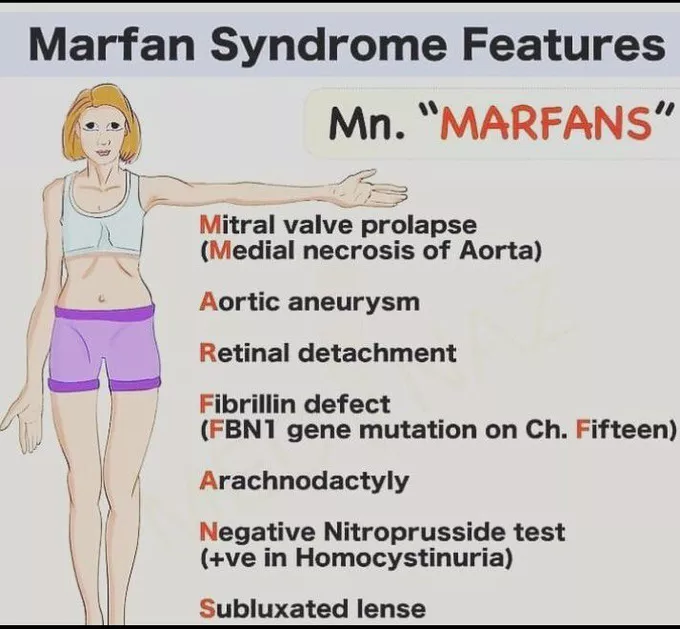
Marfan syndrome
What is Marfan syndrome?
Marfan syndrome is an inherited condition that affects connective tissue, or the fibers that support and anchor your organs and other body structures. This condition mainly affects the eyes, heart, blood vessels, and skeleton.
Marfan syndrome patients are typically tall and thin, with unusually long legs, arms, fingers, and toes. Marfan syndrome can cause either mild or severe injury. If your aorta, the large blood vessel that transports blood from your heart to the rest of your body, is involved, the disease can become fatal.
Treatment usually consists of drugs that keep your blood pressure low in order to reduce the strain on your aorta. It is critical to inspect for damage progression on a regular basis. Many patients with Marfan syndrome eventually require aortic repair surgery.
Symptoms of Marfan syndrome
Occasionally Marfan syndrome is so mild, that rare if any, symptoms are observable right away. In most patients, symptoms become obvious as changes in connective tissue occur as you age.
Because Marfan syndrome involves your connective tissue, it can involve your entire body involving your skeletal system, eyes, heart and blood vessels, skin, and organs.
Physical appearance
Physical features contain:
- A long, narrow face.
- Tall and thin body build.
- Arms, fingers, legs, and toes may appear too long for the rest of your body.
- Curved spine. Scoliosis involves 60% of patients with Marfan syndrome.
- Sternum (Breastbone) may either stick out or be indented.
- Joints that are weak and easily evolved dislocated.
- Flat feet.
Dental issues
Dental problems contain:
- Crowded teeth.
- Narrow, higher than usually arched palate (roof of the mouth).
Eye issues
More than half of all patients with Marfan syndrome have eye issues.
These contain:
- Nearsightedness (blurring of things far away).
- Lens subluxation (the lens of the eye shifts away from its usual position).
- Cataracts.
- A dissimilarity in the shape of the eye.
- Retinal detachment.
- Glaucoma.
Heart and blood vessel problems
Approximately 90% of patients with Marfan syndrome create changes in their heart and blood vessels. Modifications that can develop contain:
Aortic aneurysm. The walls of the aorta, the primary artery that brings blood from the heart to the rest of your body, evolve weakly, bulge out, and could rupture (burst). This occurs most generally at the aortic root means the point where the aortic artery leaves the heart.
Aortic dissection. This is a tear in the internal layer of the three wall layers of the aorta. The tear permits blood to enter the middle layer, which expands the tear and leads to an additional separation and maybe wall rupture. This can be destructive.
Heart valve problems. Marfan syndrome can generate valve tissue to evolve weak and stretch. This conducts to valves that don’t close tightly, generating leaks and backflow of blood. The heart frequently has to work harder when valves aren’t working appropriately. The mitral valve is generally involved.
Enlarged heart. The heart muscle may broaden and weaken over time, generating cardiomyopathy, even if the heart valves are not leaking. The illness may advance to heart failure.
Abnormal heart rhythm. Arrhythmia can happen in some patients with Marfan syndrome. It’s frequently related to mitral valve prolapse.
Brain aneurysms. Patients with Marfan may have a record of intracranial (inside the skull) bleeding from a ruptured brain aneurysm.
Lung changes
The changes in lung tissue that happen with Marfan syndrome raise the risk for:
- Asthma.
- Emphysema.
- Chronic obstructive pulmonary disease (COPD).
- Bronchitis.
- Pneumonia.
- Collapsed lung (pneumothorax).
Skin changes
Skin can evolve slightly stretchy, generating stretch marks to happen, even without weight changes.
Causes of Marfan syndrome
When you have Marfan syndrome, there is a fault in the gene that encodes the form of fibrillin and the elastic fibers, a main part of connective tissue. This gene is known as fibrillin-1 or FBN1.
In most circumstances, Marfan syndrome is inherited. The design is known as autosomal dominant, indicating it happens equally in males and females and can be inherited from only one parent with Marfan syndrome. A patient who has Marfan syndrome has a 50% possibility of passing along the disease to per of their kids.
In 25% of patients, a new gene defect happens due to an unknown reason. Marfan syndrome is also guided to as an uneven expression genetic disease because not everyone with Marfan syndrome has the same symptoms and the symptoms may be worse in some people than others.
Marfan syndrome is currently born. Yet, you may not be analyzed until you’re a teen or a young grown-up.
Risk factors
Marfan syndrome involves males and females equally and happens among all races and ethnic groups. Because it’s a genetic disease, the most significant risk factor for Marfan syndrome is having a parent with the disease.
Diagnosis
Marfan syndrome can be challenging for doctors to analyze because many connective tissue diseases have equal signs and symptoms. Actually, among members of the exact family, the signs and symptoms of Marfan syndrome differ widely both in their characteristics and in their severity.
Specific combinations of symptoms and family records must be present to verify a diagnosis of Marfan syndrome. In some patients, a patient may have some characteristics of Marfan syndrome, but not sufficiently of them to be analyzed with the disease.
Heart tests
If your doctor doubts Marfan syndrome, one of the first examinations he or she may suggest is an echocardiogram. This examination uses sound waves to catch real-time pictures of your heart in motion. It studies the state of your heart valves and the size of your aorta. Further heart-imaging choices contain computerized tomography (CT) scans and magnetic resonance imaging (MRI).
If you are analyzed with Marfan syndrome, you’ll require to have regular imaging examinations to monitor the size and state of your aorta.
Eye tests
Eye examinations that may be required contain:
Slit-lamp examination. This examination checks for lens dislocation, cataracts, or a detached retina. Your eyes will require to be fully dilated with drops for this examination.
Eye pressure examination. To study for glaucoma, your eye doctor may estimate the pressure inside your eyeball by touching it with a particular instrument. Numbing eyedrops are generally used before this examination.
Genetic testing
Genetic testing is frequently used to verify the diagnosis of Marfan syndrome. If a Marfan mutation is located, family members can be tried to see if they are also involved. You may want to speak to a genetic counselor before beginning a family, to see what your possibilities are of passing on Marfan syndrome to your future kids.
Treatment of Marfan syndrome
While there is no treatment for Marfan syndrome, therapy concentrates on controlling the different difficulties of the condition. To accomplish this, you’ll require to be inspected regularly for signs that the injury caused by the condition is progressing.
In the past, a patient who had Marfan syndrome frequently died young. With frequent monitoring and modern therapy, most patients with Marfan syndrome can now predict to live a more normal life span.
Medications
Doctors frequently define blood pressure-lowering medicines to assist to control the aorta from enlarging and to decrease the risk of dissection and rupture.
Therapy
The vision issues associated with a dislocated lens in your eye frequently can be fixed with glasses or contact lenses.
Surgical
Ascending aortic root aneurysm procedure
Relying on your signs and symptoms, processes might contain:
Aortic repair. If your aorta’s diameter gets approximately 2 inches (50 millimeters) or if it widens rapidly, your doctor may suggest an operation to substitute a part of your aorta with a tube made of synthetic material. This can assist to control a life-threatening rupture. Your aortic valve may require to be substituted as well.
Scoliosis treatment. When there is notable scoliosis, a talk with a spine expert is required. Bracing and surgery are required in some patients.
Breastbone corrections. Surgical choices are available to fix the appearance of a sunken or protruding breastbone. Because these operations are frequently believed to be for cosmetic goals, your insurance might not protect the expenses.
Eye surgeries. If regions of your retina have torn or come lax from the back of your eye, surgical repair is generally successful. If you have cataracts, your clouded lens can be substituted with an artificial lens.
Living with Marfan syndrome
The expected lifespan of a patient with Marfan syndrome used to be near the mid-40s age group but now expands into the 70s which is similar to the general residents.
This improved lifespan is mostly due to:
- enhanced awareness of Marfan syndrome among doctors or health professionals
- early diagnosis
- advances in medical management and drug
- enhanced and more useful surgical methods
- proper lifestyle changes.
Lifestyle changes
If you have Marfan syndrome, it is necessary to be well-informed regarding the disease and how it can be controlled. General well-being and quality of life for people with Marfan syndrome can be enhanced by making lifestyle options that decrease the stress on their bodies. A healthcare professional can recommend proper lifestyle suggestions.
Relying on which body regions are involved and to what degree, such changes may contain:
avoiding contact sports
avoiding physical activities that rely on isometric exercises, like weight training or carrying heavy things
doing only mild, moderate activity
having specialized counseling. A female who has a widened aorta needs specialized counseling regarding the higher risks included when considering a pregnancy, due to the associated strain on this primary artery and the risk of rupture, especially during childbirth. Cardiac assessment pre-pregnancy and frequently throughout pregnancy is suggested.
Complications
- Aneurysm at the aortic root
- Aortic aneurysm and aortic dissection
- Lens dislocation
- Retinal detachment
- Scoliosis
- Chest abnormalities
Because Marfan syndrome can involve almost any portion of your body, it may generate a broad variety of difficulties.
Cardiovascular complications
The most dangerous difficulties of Marfan syndrome include the heart and blood vessels. Defective connective tissue can weaken the aorta the big artery that derives from the heart and delivers blood to the body.
Aortic aneurysm. The pressure of blood exiting your heart can cause the wall of your aorta to bulge out, such as a weak spot in a tire. In a person who has Marfan syndrome, this is most probably to occur at the aortic root where the artery exits your heart.
Aortic dissection. The wall of the aorta is created up of layers. Dissection happens when a little tear in the innermost layer of the aorta’s wall permits blood to press between the inner and outer layers of the wall. This can generate extreme pain in the chest or back. An aortic dissection weakens the vessel’s structure and can result in a rupture, which may be destructive.
Valve malformations. A patient who has Marfan syndrome can have weak tissue in their heart valves. This can create stretching of the valve tissue and abnormal valve role. When heart valves don’t function correctly, your heart frequently has to work harder to recompense. This can ultimately conduct to heart failure.
Eye complications
Eye difficulties may contain:
Lens dislocation. The focusing lens within your eye can move out of position if its verifying structures weaken. The medical word for this issue is ectopia lentis, and it happens in more than half the patients who have Marfan syndrome.
Retinal problems. Marfan syndrome also raises the complication of a detachment or tear in the retina, the light-sensitive tissue that covers the back wall of your eye.
Early-onset glaucoma or cataracts. A patient who has this syndrome tends to create these eye issues at a younger age. Glaucoma generates pressure within the eye to rise, which can injure the optic nerve. Cataracts are cloudy regions in the eye’s generally clear lens.
Skeletal complications
This syndrome raises the risk of abnormal curves in the spine, like scoliosis. It can also interrupt the normal growth of the ribs, which can generate the breastbone to either protrude or seem sunken into the chest. Foot pain and low back pain are familiar with Marfan syndrome.
Complications of pregnancy
Marfan syndrome can weaken the walls of the aorta, the major artery that exits the heart. During pregnancy, the heart pumps more additional blood than normal. This can place additional stress on the aorta, which raises the risk of a fatal dissection or rupture.
How to Prevent Marfan syndrome?
There’s no method to control it.
If a patient has this illness or knows they have a modification in the FBN1 gene, they can pursue genetic counseling to examine the risk of having a kid with the syndrome.
Though some 25 percent of patients are not inherited, specialists think these cases occur from conception. Nobody a parent or person can control the genetic change.
But the patient can decrease the risk of severe difficulties by seeking medical assets for issues and following regular checkups as a doctor suggests.
FAQ
At what age does Marfan syndrome appear?
Marfan syndrome is a congenital disease, indicating a patient has it from birth. Physical signs are occasionally present in infancy but more frequently show up after childhood or adolescence. Marfan syndrome involves about 200,000 patients in the United States, both males and females of any race or ethnic group may be involved.
Can Marfan syndrome be cured?
Although there is no treatment for Marfan syndrome, doctors use therapies to ease symptoms and control additional issues or difficulties. Treatment relies on the region of the body involved in the syndrome and may contain: Drugs to assist manage pain and issues with your heart.
How long can you live with Marfan syndrome?
The typical lifespan of a patient with Marfan syndrome used to be about the mid-40s age group but now expands into the 70s which is similar to the general residents.
Is Marfan syndrome harmful?
Marfan syndrome is a severe, potentially life-threatening disease, and an early, accurate diagnosis is important for patients with Marfan syndrome. Both clinical and genetic testing may be used to assist diagnosis of Marfan syndrome.
Should people with Marfan’s have children?
About Marfan syndrome and pregnancy, the two main problems are the risk of transmission of Marfan syndrome to the fetus and the chance of cardiovascular complications in an involved mother. The risk of transmission to the children is at least 50%, with the option of a more extreme clinical presentation.

6 Comments