
Sickle cell anemia
What is Sickle cell anemia?
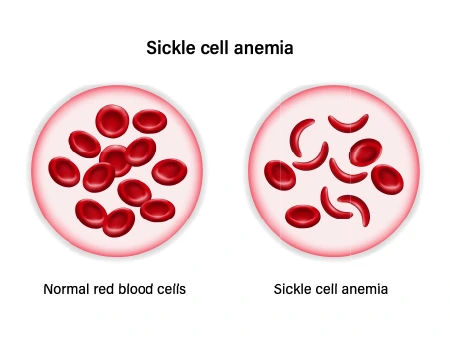
Sickle cell anemia is a form of sickle cell disorder that is inherited. Sickle cell anemia changes your red blood cells from round, flexible discs to rigid, sticky sickled cells. Sickled cells prevent red blood cells from doing their task of transporting oxygen throughout the body. Sickled cells also have a shorter lifespan than regular red blood cells. Consequently, you develop anemia, which is how sickle cell anemia got its name because you don’t have enough healthy red blood cells.
Previously, babies born with sickle cell anemia rarely survived to adulthood. Because of early detection and new treatments, approximately half of all people with sickle cell anemia now survive into their 50s. Still, sickle cell anemia patients face medical problems that could endanger their lives.
Healthcare providers, on the other hand, have treatments that reduce the chance of complications and alleviate symptoms when they occur. (Unfortunately, sickle cell anemia patients still lack access to adequate medical care in a lot of places around the world.)
How does sickle cell anemia affect a patient?
Infants born with sickle cell anemia may not include symptoms for several months. When they accomplish, symptoms contain severe tiredness or fussiness from anemia, badly swollen hands and feet, and jaundice. Additionally, infants may suffer from spleen injury, which affects the immune system and increases the likelihood of bacterial infections. As a patient with sickle cell anemia grows more aged, they may create various and more severe medical issues that occur when organ tissues don’t obtain sufficient oxygen. Patients with sickle cell anemia are at raised risk for stroke and kidney, lung, spleen, and liver injury.
What are the types of sickle cell disease?
Hemoglobin is a protein found in red blood vessels that transports oxygen. It generally has two alpha chains and two beta chains. The four main varieties of sickle cell anemia are generated by various mutations in these genes.
Hemoglobin SS disease
Hemoglobin SS condition is the most familiar variety of sickle cell disease. It happens when you inherit copies of the hemoglobin S gene from both parents. This includes hemoglobin called Hb SS. As the most extreme form of SCD, people with this form also feel the worst symptoms at a more elevated rate.
Hemoglobin SC disease
Hemoglobin SC condition is the second most familiar variety of sickle cell disease. It happens when you inherit the Hb C gene from one parent and the Hb S gene from the further. People with Hb SC have equal symptoms to people with Hb SS. Yet, the anemia is less extreme.
Hemoglobin SB+ (beta) thalassemia
Hemoglobin SB+ (beta) thalassemia involves beta globin gene production. The dimension of the red blood cell is decreased because less beta protein is produced. If inherited with the Hb S gene, you will include hemoglobin S beta-thalassemia. Symptoms are not as extreme.
Hemoglobin SB 0 (Beta-zero) thalassemia
Sickle beta-zero thalassemia is the fourth variety of sickle cell diseases. It also includes the beta-globin gene. It has equal symptoms to Hb SS anemia. Yet, occasionally the symptoms of beta zero thalassemias are more extreme. It is associated with a poorer prediction.
Hemoglobin SD, hemoglobin SE, and hemoglobin SO
These varieties of sickle cell disease are more irregular and generally, don’t have extreme symptoms.
Sickle cell trait
A patient who only inherits a mutated gene (hemoglobin S) from one parent is said to have sickle cell trait. They may have no symptoms or decreased symptoms.
Symptoms of Sickle cell anemia
Signs and symptoms of sickle cell anemia generally seem near 6 months of age. They differ from person to person and may alter over time. Signs and symptoms can contain:
Anemia. Sickle cells break separated easily and die. Red blood cells generally live for approximately 120 days before they require to be replaced. But sickle cells generally die in 10 to 20 days, leaving a lack of red blood cells (anemia). Without sufficiently red blood cells, the body can’t get sufficient oxygen and this generates fatigue.
Episodes of pain. Periodic episodes of severe pain, known as pain problems, are a primary symptom of sickle cell anemia. When red blood cells with a sickle shape prevent blood from flowing through tiny blood vessels to your joints, chest, and abdomen, pain results.
The pain differs in intensity and can last for a few hours to a few days. Some patients have only a few pain problems a year. Further, have a dozen or more a year. An extreme pain problem needs a hospital stay.
In addition, some adults and adolescents with sickle cell anemia suffer from chronic pain, which may be brought on by injuries to the bones and joints, ulcers, or other conditions.
Swelling of hands and feet. The swelling is generated by sickle-shaped red blood cells clogging blood circulation in the hands and feet.
Frequent infections. Sickle cells can injure the spleen, raising exposure to infections. Children with sickle cell anemia generally get vaccinations and antibiotics to manage potentially life-threatening infections, such as pneumonia.
They delayed growth or puberty. Red blood cells deliver the body the oxygen and nutrients required for growth. A lack of healthy red blood cells can delay development in babies and kids and slow puberty in teenagers.
Vision problems. Tiny blood vessels that provide the eyes can evolve clogged with sickle cells. This can injure the retina the part of the eye that processes visual images and conduct to vision issues.
Causes of Sickle cell anemia
Sickle cell paleness is produced by an adjustment of the quality which implies the body makes the iron-rich blend in red platelets known as hemoglobin. Hemoglobin helps red blood cells to bring oxygen from the lungs throughout the body. The hemoglobin associated with sickle cell anemia generates red blood cells to evolve rigid, moist, and deformed.
Both the mother and father must bring one copy of the sickle cell gene—also known as the sickle cell trait—and give the child both documents in the modified form for the child to participate.
If only one parent gives the sickle cell gene to the kid, that kid will have the sickle cell trait. With one specific hemoglobin gene and one modified form of the gene, patients with the sickle cell trait make both specific hemoglobin and sickle cell hemoglobin.
Their blood might include some sickle cells, but they commonly don’t have symptoms. They’re carriers of the condition, yet, which suggests they can pass the gene to their kids.
Risk factors
Kids are only at risk for sickle cell anemia if both parents bring the sickle cell trait. A blood examination known as hemoglobin electrophoresis can also specify which kind you might bring.
Patients from areas that have endemic malaria are more probable to be carriers. This contains patients from:
- Africa
- India
- the Mediterranean
- Saudi Arabia
Diagnosis
All babies in the United States are strained for sickle cell anemia. Prebirth testing examines the sickle cell gene in your amniotic fluid.
In kids and grown-ups, one or more of the subsequent methods may also be used to analyze sickle cell disease.
Detailed patient history
This disease frequently first seems as acute pain in the hands and feet. Persons may also have:
- extreme pain in the bones
- anemia
- painful enlargement of the spleen
- growth issues
- respiratory infections
- ulcers of the legs
- heart issues
Your doctor may want to examine you for sickle cell anemia if you have any of the symptoms noted above.
Blood tests
Several blood examinations can be used to look for SCD:
Blood counts can show an abnormal Hb level in the range of 6 to 8 grams per deciliter.
Blood films may conduct RBCs that seem like irregularly contracted cells.
Sickle solubility examinations look for the actuality of Hb S.
Hb electrophoresis
Hb electrophoresis is always required to verify the diagnosis of sickle cell disease. It estimates the different varieties of hemoglobin in the blood.
Treatment of Sickle cell anemia
Management of sickle cell anemia is generally aimed at avoiding pain attacks, reducing symptoms, and controlling difficulties. Treatments might contain drugs and blood transfusions. For some kids and teenagers, a stem cell transplant might cure the condition.
Medications
Hydroxyurea (Hydrea, Droxia, Siklos). Everyday hydroxyurea decreases the frequency of painful problems and might decrease the requirement for blood transfusions and hospitalizations. But it can raise the risk of infections. Don’t carry the medicine if you’re pregnant.
L-glutamine oral powder (Endari). The FDA just authorized this medicine for the therapy of sickle cell anemia. It assists in decreasing the frequency of pain crises.
Crizanlizumab (Adakveo). This medicine, provided by injection, can assist decrease the frequency of pain crises in grown-ups and kids more senior than 16. Side effects can contain joint pain, nausea, back pain, and fever.
Voxelotor (Oxbryta). This medicine is used to manage sickle cell disease in grown-ups and kids older than 12. Taken orally, this medicine can lower the risk of anemia and enhance blood flow throughout the body. Side effects can contain nausea, headache, fatigue, diarrhea, rash, and fever.
Pain-relieving medications. Your doctor might define narcotics to assist in reducing pain during sickle cell pain problems.
Preventing infections
Kids with sickle cell anemia might obtain penicillin between the ages of approximately 2 months old and at least age 5 years. As a result, infections like pneumonia, which can be fatal for children with sickle cell anemia, can be controlled.
Grown-ups who have sickle cell anemia might require to bring penicillin throughout their lives if they’ve had pneumonia or surgery to extract the spleen.
Preadolescence vaccinations are essential for controlling conditions in all kids. They’re even more essential for kids with sickle cell anemia because their infections can be severe.
Your kid’s doctor should confirm that your kid obtains all the suggested childhood vaccinations, as well as vaccines against meningitis, pneumonia, hepatitis B, and an annual flu shot. Vaccines are also essential for grown-ups with sickle cell anemia.
During the COVID-19 pandemic, patients with sickle cell anemia should bring additional precautions, like remaining isolated at home as much as likely and for those who are qualified, obtaining vaccinated.
Surgical and other procedures
Blood transfusions. These are used to manage and control difficulties, like stroke, in a patient with sickle cell disease.
In a red blood cell transfusion, red blood cells are extracted from a collection of donated blood, then given via a vein to people with sickle cell anemia. This raises the digit of normal red blood cells, which assists to decrease symptoms and difficulties.
Risks contain an immune reaction to the donor blood, which can create it difficult to find future donors, infection, and extra iron buildup in your body. Because extra iron can injure your heart, liver, and further organs, you might require therapy to decrease iron levels if you undergo routine transfusions.
Stem cell transplant. Also called a bone marrow transplant, this method includes replacing bone marrow involved by sickle cell anemia with healthy bone marrow from a donor. The method generally uses a matched donor, like a sibling, who doesn’t have sickle cell anemia.
Because of the risks associated with a bone marrow transplant, involving death, the method is suggested only for the patient, generally, kids, who have significant symptoms and difficulties of sickle cell anemia. A stem cell transplant is an only known treatment for sickle cell anemia.
Clinical tests are ongoing to manage stem cell transplantation in grown-ups and gene treatments.
Lifestyle and Home remedies
Bringing the subsequent steps to remain healthy might assist you to avoid the difficulties of sickle cell anemia:
Carry folic acid supplements daily and select a healthy diet. Bone marrow requires folic acid and further vitamins to create new red blood cells. Ask your doctor regarding a folic acid supplement and further vitamins. Consume a mixture of colorful fruits and vegetables, as well as whole grains.
Drink plenty of water. Dehydration can raise your risk of a sickle cell crisis. Consume water throughout your day, purposing for about eight glasses a day. Raise the quantity of water you consume if you exercise or consume time in a hot, dry climate.
Avoid temperature extremes. Exposure to excessive heat or cold can raise your risk of a sickle cell crisis.
Exercise daily, but don’t exaggerate it. Speak with your doctor regarding how much activity is right for you.
Use nonprescription drugs with caution. Use pain drugs, like ibuprofen (Motrin IB, Advil, Children’s Motrin, others) or naproxen sodium (Aleve), sparingly, if at all, because of the possible impact on your kidneys. Ask your doctor before bringing nonprescription medicines.
Don’t smoke. Smoking raises your risk of pain problems.
Complications of Sickle cell anemia
Sickle cell anemia can conduct to a host of difficulties, involving:
Stroke. Sickle cells can stop blood flow to a region of the brain. Signs of stroke involve seizures, weakness or numbness of the arms and legs, sudden speech problems, and failure of consciousness. If your kid has any of these signs and symptoms, pursue medical treatment instantly. A stroke can be fatal.
Acute chest syndrome. Chest pain, fever, and difficulty breathing can be caused by a lung infection or sickle cell disease that clogs blood vessels in the lungs. It might need emergency medical therapy.
Pulmonary hypertension. Patients with sickle cell anemia can produce high blood pressure in their lungs. This difficulty generally involves grown-ups. Shortness of breath and fatigue are familiar symptoms of this disease, which can be fatal.
Organ damage. Sickle cells that clog blood flow to organs deprive the involved organs of blood and oxygen. In sickle cell anemia, blood is also chronically down in oxygen. This absence of oxygen-rich blood can injure nerves and organs, involving the liver, kidneys, and spleen, and can be fatal.
Splenic sequestration. A big number of sickle cells can obtain trapped in the spleen, generating it to broaden and possibly generating belly pain on the left side of the body. This can exist life-threatening. Parents of kids with sickle cell anemia should understand to regularly feel their kid’s spleen for enlargement.
Blindness. Sickle cells can clog small blood vessels that supply the eyes. Over time, this can conduct to blindness.
Leg ulcers. Sickle cell anemia can generate painful open sores on the legs.
Gallstones. The breakdown of red blood cells creates a substance known as bilirubin. A high level of bilirubin in the body can conduct gallstones.
Priapism. In this disease, males with sickle cell anemia can have painful, long-lasting erections. Sickle cells can clog the blood vessels in the penis, which can conduct to impotence over a period.
Deep vein thrombosis. Red cell sickling can lead to the formation of blood clots, which can increase the likelihood of a clot forming in a deep vein (deep vein thrombosis) or lung (pulmonary embolism). Either can generate severe disease or even death.
Pregnancy difficulties. Sickle cell anemia can raise the risk of high blood pressure and blood clumps during pregnancy. It can also raise the risk of miscarriage, premature birth, and delivering low birth weight infants.
Prevention
Seeing a geneticist before trying to conceive if you have the sickle cell trait can help you understand your risk of having a sickle cell anemic child. A genetic consultant can also describe possible therapies, preventive actions, and reproductive choices.
Prognosis
In history, patients with sickle cell disease frequently died between the ages of 20 and 40. Thanks to contemporary care, the patient now can live to the age of 50 and beyond.
The reasons for death contain organ failure and infection.
FAQ
What occurs to a patient with sickle cell anemia?
Sickle cells that obstruct blood flow to organs deprive the involved organs of blood and oxygen. In sickle cell anemia, blood is also chronically down in oxygen. This absence of oxygen-rich blood can injure nerves and organs, involving the liver, kidneys, and spleen, and can be fatal.
Is sickle cell a serious disease?
Patients with sickle cell conditions create unusually shaped red blood cells that can generate issues because they do not live as long as nutritional blood cells and can clog blood vessels. Sickle cell disease is a severe and lifelong health disease, although therapy can assist to control many of the symptoms.
Can sickle cell anemia be cured?
A blood and bone marrow transplant is presently the only treatment for some people who have sickle cell disease. After early diagnosis, your healthcare provider may suggest drugs or transfusions control difficulties, involving chronic pain.
How many years do sickle cell patients live?
Low Life Expectancy with Sickle Cell Disease
Therefore, their life expectancy is decreased compared with that of the general people. According to a recent study, adults with SCD have a life expectancy of 54 years, which is approximately 20 years less than that of normal adults without SCD.
Is sickle cell Painful?
Pain is the most common problem for SCD patients and the most common reason they visit the hospital or emergency room. Sickled cells traveling via small blood vessels can bring stuck and block blood flow throughout the body, generating pain.



One Comment