
CYSTIC FIBROSIS
DEFINATION – CYSTIC FIBROSIS(CF) is a hereditary disorder of the exocrine glands, with a high sodium chloride content in sweat and pancreatic insufficiency, resulting in the, malabsorption . There is hypertrophy and hyperplasia of mucus secreting glands, resulting in the excessive mucus production in the lining if the bronchi , which predispose the patient to chronic bronchopulmonary infection”.
Cystic fibrosis is an inherited disorder that causes severe damage to the lungs, digestive system and other organs in the body.
Cystic fibrosis affects the cells that produce mucus, sweat and digestive juices. These secreted fluids are normally thin and slippery. But in people with cystic fibrosis, a defective gene causes the secretions to become sticky and thick. Instead of acting as a lubricant, the secretions plug up tubes, ducts and passageways, especially in the lungs and pancreas.
Although cystic fibrosis requires daily care, people with the condition are usually able to attend school and work, and often have a better quality of life than people with cystic fibrosis had in previous decades. Improvements in screening and treatments mean people with cystic fibrosis now may live into their mid- to late 30s, on average, and some are living into their 40s and 50s.
SYMPTOMS-
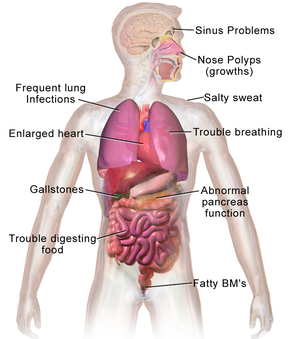
Cystic fibrosis signs and symptoms vary, depending on the severity of the disease. Even in the same person, symptoms may worsen or improve as time passes. Some people may not experience symptoms until adolescence or adulthood.
People with cystic fibrosis have a higher than normal level of salt in their sweat. Parents often can taste the salt when they kiss their children. Most of the other signs and symptoms of cystic fibrosis affect the respiratory system and digestive system. However, adults diagnosed with cystic fibrosis are more likely to have atypical symptoms, such as recurring bouts of inflamed pancreas (pancreatitis), infertility and recurring pneumonia.
RESPIRATORY SIGNS AND SYMPTOMS-
The thick and sticky mucus associated with cystic fibrosis clogs the tubes that carry air in and out of your lungs. This can cause signs and symptoms such as:
- A persistent cough that produces thick mucus (sputum)
- Wheezing
- Breathlessness
- Exercise intolerance
- Repeated lung infections
- Inflamed nasal passages or a stuffy nose
DIGESTIVE SIGNS AND SYMPTOMS-
The thick mucus can also block tubes that carry digestive enzymes from your pancreas to your small intestine. Without these digestive enzymes, your intestines aren’t able to completely absorb the nutrients in the food you eat. The result is often:
- Foul-smelling, greasy stools
- Poor weight gain and growth
- Intestinal blockage, particularly in newborns (meconium ileus)
- Severe constipation
Frequent straining while passing stool can cause part of the rectum — the end of the large intestine , to protrude outside the anus (rectal prolapse). When this occurs in children, it may be a sign of cystic fibrosis. Parents should consult a physician knowledgeable about cystic fibrosis. Rectal prolapse in children may sometimes require surgery. Rectal prolapse in children with cystic fibrosis is less common than it was in the past, which may be due to earlier testing, diagnosis and treatment of cystic fibrosis.
CAUSES AND RISK FACTORS-
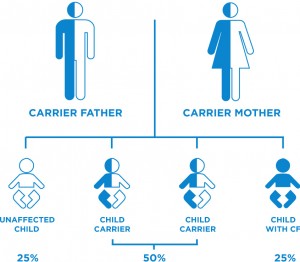
Cystic Fibrosis is an autosomal recessive disease caused by defects in the CFTR gene . This CFTR gene normally encodes for a protein that functions as a chloride channel and is regulated by cyclic AMP, but in cystic fibrosis, the CFTR gene mutates and results in abnormalities of cyclic AMP, which regulates the transport of chloride across the epithelial cells on mucosal surfaces. This defective gene causes the body to produce an abnormally thick and sticky fluid in the respiratory and GI tracts, the pancreas and in the sweat glands.
In order to develop CF, two CF genes must be inherited, one from the mother and the other from the father. If only one CF gene is inherited, then the person is called a carrier and will have no symptoms. When both parents are carriers, with each pregnancy there is a: 1 in 4 chance of having a child with cystic fibrosis, 1 in 2 chance of having a child who is a carrier or 1 in 4 chance of having an unaffected child.
There are several different types of genetic mutation which are associated with different degrees of severity of the disease
- COMPLICATIONS-
RESPIRATORY SYSTEM AND COMPLICATIONS-
1.Damaged airways (bronchiectasis). Cystic fibrosis is one of the leading causes of bronchiectasis, a condition that damages the airways. This makes it harder to move air in and out of the lungs and clear mucus from the airways (bronchial tubes). - 2.Chronic infections. Thick mucus in the lungs and sinuses provides an ideal breeding ground for bacteria and fungi. People with cystic fibrosis may often have sinus infections, bronchitis or pneumonia.
- 3.Growths in the nose (nasal polyps). Because the lining inside the nose is inflamed and swollen, it can develop soft, fleshy growths (polyps).
- 4.Coughing up blood (hemoptysis). Over time, cystic fibrosis can cause thinning of the airway walls. As a result, teenagers and adults with cystic fibrosis may cough up blood.
- 5.Pneumothorax. This condition, in which air collects in the space that separates the lungs from the chest wall, also is more common in older people with cystic fibrosis. Pneumothorax can cause chest pain and breathlessness.
- 6.Respiratory failure. Over time, cystic fibrosis can damage lung tissue so badly that it no longer works. Lung function usually worsens gradually, and it eventually can become life-threatening.
- 7.Acute exacerbations. People with cystic fibrosis may experience worsening of their respiratory symptoms, such as coughing and shortness of breath, for several days to weeks. This is called an acute exacerbation and requires treatment in the hospital.
- DIGESTIVE SYSTEM AND COMPLICATIONS-
- 1.Nutritional deficiencies. Thick mucus can block the tubes that carry digestive enzymes from your pancreas to your intestines. Without these enzymes, your body can’t absorb protein, fats or fat-soluble vitamins.
- 2.Diabetes. The pancreas produces insulin, which your body needs to use sugar. Cystic fibrosis increases the risk of diabetes. Around 30 percent of people with cystic fibrosis develop diabetes by age 30.
- 3.Blocked bile duct. The tube that carries bile from your liver and gallbladder to your small intestine may become blocked and inflamed, leading to liver problems and sometimes gallstones.
- 4.Intestinal obstruction. Intestinal obstruction can happen to people with cystic fibrosis at all ages. Children and adults with cystic fibrosis are more likely than are infants to develop intussusception, a condition in which a section of the intestines folds in on itself like an accordion.
- 5.Distal intestinal obstruction syndrome (DIOS). DIOS is partial or complete obstruction where the small intestine meets the large intestine.
- REPRODUCTIVE SYSTEM AND COMPLICATIONS-
1.Almost all MEN with cystic fibrosis are infertile because the tube that connects the testes and prostate gland (vas deferens) is either blocked with mucus or missing entirely. Certain fertility treatments and surgical procedures sometimes make it possible for men with cystic fibrosis to become biological fathers. - 2.Although WOMEN with cystic fibrosis may be less fertile than other women, it’s possible for them to conceive and to have successful pregnancies. Still, pregnancy can worsen the signs and symptoms of cystic fibrosis, so be sure to discuss the possible risks with your doctor.
OTHER COMPLICATIONS-
1.Thinning of the bones (osteoporosis). People with cystic fibrosis are at higher risk of developing a dangerous thinning of bones.
2.Electrolyte imbalances and dehydration. Because people with cystic fibrosis have saltier sweat, the balance of minerals in their blood may be upset. Signs and symptoms include increased heart rate, fatigue, weakness and low blood pressure.
DIAGNOSIS-
With a complete medical history and physical examination, diagnostic procedure may include the following:
SWEAT (CHLORIDE)TEST: A test, which measures the amount of chloride in the sweat. If the amount of chloride is higher than the normal amounts it may suggest cystic fobrosis. The sweat test is not painful.
1.BLOOD TEST: Blood or cheek scraping cells can also be used to detect mutations of the CFTR gene.
2.CHEST X-RAY: A diagnostic test, which produce images of internal tissues, bones and organs.
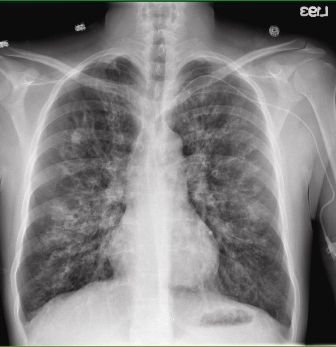
cystic-fibrosiss3.PULMONARY FUNCTION TEST: Measures the lungs’ ability to exhange oxygen and carbondioxide appropriately.
4.SPUTUM CULTURE: Is often performed to determine if an infection is present.
5.STOOL EVACUATION-: To measure stool fat absorption.Pancreatic function
6.COMPUTED TOMOGRAPHY SCAN: Detect early pulmonary disease before synptoms occur.
7.MAGNETIC RESONANCE IMAGINE(MRI): Is not only used to screen the lung, but also the heart, sinuses and GI tract.
PROGNOSIS-
Prognosis has improved over the past fifty years because of advancement in the management, especially before the irreversible pulmonary changes. The median age for survival is 35 years, long term survival is better in patients without pancreatic insufficiency. The following have been associated with worse prognosis; females, early colonization with mucoid Pseudomonas, presentation with respiratory symptoms, tobacco smoke exposure and airway hyperactivity.
MEDICAL MANAGEMENT-
Although airway clearance continues to be an integral part of care, physical exercise, postural care and the need to address the unique complications which emerge as a result of improved longevity have significantly changed the nature of physiotherapy in CF.
There is no cure for CF and most people with the disease die between the ages of 20 and 30 as a result of lung failure. Early diagnosis and a complete treatment plan can improve both survival and quality of life for the person suffering from CF.
MEDICAL MANAGEMENT-
Treatment of CF includes:
Antibiotics for respiratory infections
Vitamin supplements
Inhaled medicines to help open the airways
DNAse enzyme replacement therapy to thin the mucus and makes it easier to expectorate
Pain relievers
Enzyme therapy is done with each meal or snack, where most people with CF will need to take replacement enzymes such as pancreatin (eg Pancrex). These supply the missing pancreatic enzymes and allow proper digestion. People with CF normally need vitamin and mineral supplements too.
The usual childhood vaccinations, such as measles, mumps and rubella (MMR) and diphtheria, tetanus and whooping cough (DTP) are important for the children with CF. If you have CF you should also be vaccinated against flu and pneumococcus to help prevent chest infections.
CF is one of the most extensively researched genetic diseases as a target for gene therapy development and this can also serve as an important model for gene therapy of other diseases. The aim of gene therapy is to correct the basic defect in CF. This can be achieved by the development of a vector. This can be done by doing preclinical and clinical research. Clinical studies have identified the host immune response and low vector efficiency as key impediments to effective CF gene therapy.
PHYSIOTHERAPY MANAGEMENT-
The main aim of physiotherapy is to remove excessive mucoid secretions in order to improve ventilation. A person suffering from CF will require intensive chest physiotherapy. This will include the following:
- VIGOROUS MASSAGE to help loosen the sticky mucus
- PERCUSSION-This technique is also known as chest clapping, and is used to help loosen secretions. To perform this technique a cupped hand is used to clap the chest firmly and rhythmically (over a layer of clothing or a towel) .
- SHAKING AND VIBRATION- This technique consists of several short rhythmical squeezes to the chest while exhaling to mobilze secretions .
Regular assessment and monitoring is necessary during physiotherapy treatment as the patient may require supplemental oxygen, especially in advanced cystic fibrosis.
Other techniques include:
- POSITIONING- lying on either side, front or back and sitting
Active cycle of breathing (ACBT): This technique consist of 1.Breathing Control (BC), 2.Thoracic Expansion Exercises (TEE), and 3.Forced Expiration Technique (FET). - ACBT
1.Breathing Control (BC): Relaxed diaphragmatic breathing which allows for rest and helps avoid any tightening of airways, which can make it difficult to clear secretions
2.Thoracic expansion exercises (TEE): Deep breathing exercises that help the lungs to expand more effectively and allow air to get behind secretions so that they can be “pushed” up the airways towards the mouth. The breaths should be slow and deep with a pause at the end of inspiration then followed by relaxed quiet expiration .
3.Forced Expiration Technique (FET): This consists of huffing or a sigh, to help move secretions from the smaller to the larger airways from where they can be cleared more easily. - Positive expiratory pressure (PEP): A technique that helps to open up airways and get air behind secretions to help move them higher up the airway. The PEP device (mask or mouthpiece) gives a small degree of resistance to the breath out and this resistance splints open the airways. Bubble PEP is used in young children and involves the child blowing through a straw placed in a bottle of water and soap to produce bubbles
- Oscillating PEP- These devices combine vibrations of airways with PEP and there are several different types. The Flutter is a small pipe shaped device that interrupts the flow of air and gives an intermittent back pressure to the airways as well as causing them to vibrate. The Cornet and the Acapella are two other oscillating PEP devices that function similarily to the Flutter device
- POSTURAL DRAINAGE–
Postural drainage sounds complicated, but it’s really just a way to use gravity to drain mucus out of your lungs by changing positions. It’s used to treat a variety of conditions, including chronic diseases such as cystic fibrosis and bronchiectasis, as well as temporary infections, such as pneumonia.
If you have a bad cold or flu, you can also use postural drainage to help keep mucus out of your lungs. The goal is to move mucus into the central airway, where it can be coughed up. It’s safe for people of all ages and can be done either at home or in a hospital or nursing facility.
Postural drainage is often done at the same time as percussion, sometimes called clapping, which involves someone clapping on your back, chest, or sides with a cupped hand in order to shake mucus loose from the lungs. These techniques, along with vibration, deep breathing, and huffing and coughing, are referred to as chest physiotherapy, chest physical therapy, or airway clearance therapy.
TEQNIC OF POSTURAL DRAINAGE-
You can do postural drainage with many positions, either on your own or with a physical therapist or nurse.
- General guidelines
Each position should be held for a minimum of five minutes.
Positions can be done on a bed or on the floor.
In each position, your chest should be lower than your hips to allow mucus to drain.
Use pillows, foam wedges, and other devices to make yourself as comfortable as possible.
While in the positions, try to breathe in through your nose and out through your mouth for longer than you breathe in for maximum effectiveness.
Do these positions in the morning to clear mucus that’s built up overnight or right before bed to prevent coughing during the night.
A respiratory therapist, nurse, or doctor can recommend the best ways to perform postural drainage based on where the mucus is. - ON YOUR BACK-
Your chest should be lower than your hips, which you can achieve by lying on a slanted surface or propping your hips up about 18 to 20 inches with pillows or another item.
This position is best for draining the bottom front parts of your lungs. - ON YOUR SIDES-
With pillows under your hips, lie on one side so that your chest is lower than your hips.
To clear congestion from the bottom part of the right lung, lie on your left side.
To clear congestion from the bottom part of your left lung, lie on your right side. - ON YOUR STOMACH-
Drape your body over a stack of pillows or other object, such as a beanbag, and rest your arms by your head, with your chest lower than your hips.
This position is best for clearing mucus in the lower back area of the lungs.
More research is needed to know how effective postural drainage really is. In the meantime, your doctor may be able to suggest postural drainage positions or other chest physiotherapy techniques that may work for you. They can also refer you to a respiratory therapist or physical therapist who specializes in chest physiotherapy.
RISK ASSOCIATED POSTURAL DRAINAGE-
You may vomit if you do postural drainage right after eating. Try to do the positions before eating or 1 1/2 to 2 hours after a meal.
If left untreated, mucus in the lungs can turn into a serious condition, so make sure to follow up with your doctor if you decide to try postural drainage. You may need additional treatment. Mucus in the lungs can also be a sign of an underlying condition that needs medical treatment, such as chronic pulmonary obstructive disease (COPD).
Get emergency treatment if you have any of the following symptoms during or after postural drainage:
- Shortness of breath
- Trouble breathing
- Confusion
- Skin that turns blue
- Coughing up blood
- Severe pain
Author: Nitesh Patel - Physiotherapist
Physiotherapist in Samarpan Physiotherapy Clinic Ahmedabad Bapunagar Amaraiwadi Vastral Mobile Physiotherapy Clinic Dr. Nitesh Patel ( Physiotherapist ) : Mo No : 09898607803


8 Comments